

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Idiopathic pulmonary fibrosis (IPF) induces the proliferation of fibroblasts because of the activation due to the repeated damage of the alveolar epithelial cells. Thus, it is involved in the formation of fibroblastic foci and this increases the extracellular matrix, thus contributing to the wound healing1. Epithelial cells release procoagulant factor and TGF-β and thereby promote the migration and proliferation of fibroblasts. Due to the resulting increase in the secretion of collagen, they are involved in the pulmonary fibrosis2. In recent years, it has been reported that an epithelial-mesenchymal transition (EMT) phenomenon where the alveolar epithelial cells are directly converted into the mesothelial cells might be one of the key mechanisms by which the pulmonary fibrosis occurs. In this process, TGF-β1 plays a key role3. As another mechanism by which the pulmonary fibrosis occurs, there is a phenomenon that the plasma proteins are leaked into the alveoli due to the damage of pulmonary tissue and this activates the blood coagulation factors and then leads to the deposition of fibrin. This is primarily dissolved by plasmin, which is formed by the degradation of plasminogen with the mediation of tissue-type plasminogen activator (tPA) or urokinase-type plasminogen activator (uPA). PAI-1 binds to tPA or uPA and thereby impairs the formation of plasmin. This eventually inhibits the dissolution of fibrin and contributes to the fibrosis of various types of tissue4. As a matter of fact, PAI-1 is increasingly expressed in cases of hypertension-induced glomerulosclerosis5. Its expression is also increased in cases of carbon tetrachloride-induced liver cirrhosis6 and bleomycin-induced pulmonary fibrosis7. In type II alveolar epithelial cells of patients with idiopathic pulmonary fibrosis (IPF), the expression of PAI-1 has been reported to be increased. In particular, an animal experiment using a mouse has shown that the inhibition of PAI-1 with PAI-1 siRNA reduced the progression of pulmonary fibrosis due to bleomycin8. As described here, it is presumed that PAI-1 might play a pivotal role in the pathogenesis of pulmonary fibrosis and asthma9,10.
To date, however, no studies have closely examined the effects of PAI-1 on the EMT, one of the possible key mechanisms by which the pulmonary fibrosis occurs. We isolated Type II alveolar epithelial cells from rat and then cultured them. Then, we exposed them to the smoking extract and thereby induced the occurrence of EMT. Thus, we examined whether the PAI-1 would play a pivotal role or whether the suppression of the expression of PAI-1 would lead to that of EMT. In addition, we created an animal experimental model of bleomycin-induced pulmonary fibrosis using in rat and thereby examined the role of PAI-1.
Materials and Methods
1. Isolation and culture of type ll epithelial cells and their transfection with PAI-1 siRNA
Alveolar type II epithelial cells were isolated from the lung of SD-rat based on the density gradient11. Rats were given an intra-abdominal injection of Zoletil 50 mg/kg. Thus, the anesthesia was performed. The bronchoalveolar lavage (BAL) was performed using PBS five times. A 2.5% trypsin 5 mL was infused in the lung. Then, the lung was tied to prevent the leakage and then extracted. The sample was placed at 37℃ for 30 minutes. The tissue sample was sectioned and the trypsin was neutralized using a 5 mL FBS. The total volume was set at 20 mL using PBS, to which a 4% DNase I solution was added. Then, the reaction was performed at 4℃ for five minutes. The sample was placed on to a layer of percoll (light, 1.040: heavy, 1.090) which was already prepared. The centrifugation was done at 300 g for 20 minutes. Thus, the layer of type II epithelial cells was isolated. To prevent the contamination of fibroblasts, the isolated epithelial cells were placed in the DMEM media and then cultured in a petri-dish for an hour. Then, the supernatant was harvested for further experimental procedure.
For the PAI-1 siRNA transfection, the media of epithelial cells were converted into those where there is a lack of the antibiotics. This was followed by a 24 hours culture. The culture media were replaced by siRNA transfection media, followed by the treatment with 0.75µg PAI-1 siRNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA), which were subjected to a 6-hr transfection. Thereafter, the culture media were replaced by the normal growth media and then used for further laboratory procedures 72 hours later.
2. Cigarette smoking extract (CSE) and the treatment with tiplaxtinin
In a 25 mL DMEM medium (HEPES, 50 mM without FBS or penicillin/streptomycin), the cigarette smoke was collected for five minutes at a rate of 1 puff/min. The titration was done to create a pH of 7.4. It was filtered using a 0.22µm filter unit (Millipore, Bedford, MA, USA) and then used for the CSE. The CSE which was prepared as described herein was used for the experimental procedure within 30 minutes. Type II eithelial cells were treated with tiplaxtinin at varying concentrations (10-, 15- and 50µM) for 30 minutes. This was followed by a 72 hours stimulation of the cells with 5% CSE. A 24- and a 2-hour stimulation of the cells with CSE were done to observe the RNA and the cellular signal transduction, respectively. In addition, PAI-1 siRNA and TGF-β (5 ng/mL) were served as negative and positive controls, respectively.
3. An animal experimental model using bleomycin
In male Sprague-Dawley rats aged seven weeks, a 4 U/kg bleomycin was administered to the airway tract. After 1-, 3-, 5-, 7- and 10 days, a 1 mg/kg tiplaxtinin was orally administered. After 14 days, the anesthesia was performed using zoletil. After the BAL was performed using PBS three times, the lung tissue samples were obtained. The left lung tissue of mice was fixed in a 4% paraformaldehyde. This was followed by the preparation of paraffin-embedded blocks. After the blocks were sectioned at a thickness of 4µm, they were stained using an H&E dye. The lesions on the stained tissue samples were converted into the numerical values based on the criteria of Aschcroft score.
4. Enzyme-linked immunosorbent assay (ELISA)
The concentrations of PAI-1 and TGF-β were measured from the BAL fluid and the supernatant of Type II Epithelial cells with the use of ELISA. In measuring the concentration of TGF-β, the samples were pretreated with 1N HCl and 1.3N NaOH for further laboratory procedures. The experimental procedure was performed in accordance with the manufacturer's protocol (PAI-1: Innovative Research Inc., Southfield, MI, USA; TGF-β: R&D systems, Minneapolis, MN, USA).
5. Real time RT-PCR
With the use of TRIzol, chloroform and isopropanol, the RNA was isolated from Type II epithelial cells. This was followed by the reverse transcription using a 2-µg RNA. cDNA was treated with SYBR Green Real-Time Premix (RBC Bioscience, Chung Ho City, Taiwan), which was followed by the amplification using iQ5 cycler (Bio-Rad, Hercules, CA, USA). The followings are the sequences of primer which was used herein:
α-SMA; CGG GCT TTG CTG GTG ATG/GGT CAG GAT CCC TCT CTT GCT
E-cadherin; GGC CCA GGA GCT GAC AAA C/CCA GAG GCT GCG TCA CTT TC,
PAI-1; GAG CCA GAT TCA TCA TCA ACG/CTG CAA TGA ACA TGC TGA GG,
GAPDH; CAA CTC CCT CAA GAT TGT CAG CAA/GGC ATG GAC TGT GGT CAT GA.
In regard to the PCR conditions, the PCR was performed at 60℃ in a total of 45 cycles.
6. Western blot
Type II epithelial cells and the lung tissue samples were treated with RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% deoxycholic acid, pH 7.5), by which the protein samples were obtained. Following the protein quantification, the western blot analysis was performed using 50µg. Then, the expression of α-SMA, E-cadherin and PAI-1 was examined, where an assay was performed for p-ERK, p-SMAD2 and SNAI-1 (Snail) to observe the cellular signal transduction. All the antibodies used herein were purchased from Santa Cruz, which were diluted at a ratio of 1:400 and then used to provoke the reactions.
7. Immunocytochemical staining
E-cadherin and α-SMA were served as a specific marker for epithelial cells and myofibroblasts respectively. Then, to confirm the intracellular expression of α-SMA and PAI-1, an immunocytochemical staining was performed for each protein. The cells treated with stimulants were cultured for three days. This was followed by the fixation using a 2% paraformaldehyde at 4℃ for 20 minutes. The primary antibody (α-SMA, E-cadherin, PAI-1; Santa Cruz) was treated at a concentration ratio of 1:50 and the secondary one was used by a ratio of FITC conjugated anti-mouse IgG to PE conjugated anti-rabbit IgG of 1:1,000. Besides, to make a differentiation of the cellular nucleus, the DAPI staining was done with the use of Ultra Cruz Mounting Medium. The stained cells were examined at a magnification rate of ×200 with the use of confocal microscopy (LSM510Meta; Zeiss, Oberkochen, Germany).
8. Statistical analysis
All the data was expressed as mean±standard error (SE). An inter-group comparison was made using Mann-Whitney U test. Statistical analysis was performed using SPSS version 10.0.7 (SPSS Inc., Chicago, IL, USA), where a p-value of <0.05 was considered statistically significant.
Results
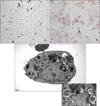
Type II epithelial cells were isolated from the lung of mice and then confirmed on an alkaline phosphatase staining. Using an electron microscopy, the lamellar body was examined (Figure 1). When the type II epithelial cells were exposed to the CSE, there were findings associated with the EMT such as the disappearance of E-cadherin mRNA and the expression of α-SMA mRNA. This was also accompanied by the increased expression of PAI-1 mRNA. These phenomena also showed that the EMT or PAI-1 mRNA were decreased by tiplaxtinin (50µM) (PAI-1 inhibitor) or the transfection with PAI-1 siRNA (Figure 2). These findings were also confirmed on a western blot analysis. To put this in another way, type II epithelial cells are associated with the increased expression of α-SMA and the decreased expression of E-cadherin. Besides, they are also associated with the increased expression of PAI-1. These findings were suppressed by the transfection with PAI-1 siRNA or tiplaxtinin (50µM) (Figure 3). An immunohistochemical staining also showed that the CSE increased the expression of α-SMA in type II epithelial cells and abolished that of E-cadherin (Figure 4). In addition, it also increased the expression of PAI-1 (Figure 5). It was observed using a confocal microscope that these findings were suppressed by the transfection with PAI-1 siRNA or tiplaxtinin (50µM).
If type II epithelial cells are converted into myoblasts by the EMT, they would release such cytokines as TGF-β and this would eventually lead to the increased synthesis of such substances as collagen. Accordingly, the concentration of TGF-β on the supernatant was measured using the ELISA. As expected, the concentration of TGF-β was increased on the supernatant of type II epithelial cells which were stimulated by the CSE. This was suppressed by the transfection with PAI-1 siRNA or tiplaxtinin (50µM) (Figure 6). Following the experiment on the signal transduction pathway due to the CSE in type II epithelial cells, there were findings that the CSE phosphorylated the SMAD2 and the expression of SNAIL was increased following the phosphorylation of ERK1/2. The expression of SNAIL due to this signal transduction was suppressed by the transfection with PAI-1 siRNA or tiplaxtinin (50µM) (Figure 7). Also in an experiment where the pulmonary fibrosis was induced by bleomycin in rats, the severe pulmonary inflammation and fibrosis were greatly suppressed by oral administration of tiplaxtinin. Also on the Ashcroft scores which were interpreted by the pathologist, the pulmonary fibrosis was found to be improved with the use of tiplaxtinin (Figure 8). In the BAL fluid of rats, the concentrations of PAI-1 and TGF-β were measured using the ELISA. The concentrations of PAI-1 and TGF-β, both of which were increased by bleomycin, were decreased with the use of tiplaxtinin (Figure 9).
Discussion
The epithelial cells in the lung are the common target sites for pulmonary damage. They play a key role in the following clinical courses with no respect to whether a normal recovery is achieved or the fibrosis occurs. In a phenomenon of the EMT where the epithelial cells lose their own characteristics and are converted into the mesenchymal cells, TGF-β is deeply involved. The resulting activation of Smad and β-catenin is also associated with the EMT12,13. Idiopathic pulmonary fibrosis (IPF) commonly occurs in smokers. But there are not a great number of studies which have been conducted to examine whether the smoking is associated with the occurrence of pulmonary fibrosis, particularly, in relation to the EMT. It has been reported that the cigarette smoking extract (CSE) triggers the occurrence of EMT at a lower concentration in A549 cell lines and it is also associated with the proliferation of myofibroblasts14. These findings suggests that it can be involved in the occurrence of idiopathic pulmonary fibrosis (IPF). In addition, CSE increased the expression of PAI-1 in A549 epithelial cells and the increased PAI-1 increased the expression of IL-8 or LTB4. This might be associated with the occurrence of inflammatory responses in patients with chronic obstructive pulmonary disease15,16. It is nowadays well established that the major functions of PAI-1 are to suppress the fibrinolysis by plasmin and thereby to contribute to the occurrence of pulmonary and renal fibrosis. In the current experimental study, however, when type II epithelial cells were isolated and then cultured and they were exposed to the CSE, the expression of PAI-1 was increased and this was directly involved in the occurrence of EMT. It was also shown that the signal transduction pathway in this process activated the SNAIL via SMAD2 and ERK1/2 and type II epithelial cells were converted into myofibroblasts accordingly. In addition, with the use of tiplaxtinin that impairs the fibrinolysis of PAI-1 due to the transfection with PAI-1 siRNA or binding with PAI-1 in epithelial cells17, the presence of this signal transduction pathway was confirmed.
Of myriads of molecules that are involved in the EMT, zinc-finger transcription factors such as SNAI1 (Snail) and SNAI2 (Slug) play a role in regulating the EMT in the diseases triggering the occurrence of cancer or fibrosis in the specific body organ18,19. In many body organs such as the lung, the most important cytokine that is involved in the EMT, TGF-β1 plays a role in migrating Snail transcription factors into the nucleus and triggering the occurrence of EMT in type II epithelial cells20. Also in the current experimental study, the CSE increased the expression of Snail transcription factors in type II epithelial cells. Following the transfection with PAI-1 siRNA or pre-treatment with tiplaxtinin, the phosphorylation of SMAD-2 and the expression of Snail transcription factor were suppressed. This was also accompanied by an EMT phenomenon that is characterized by the decreased expression of α-SMA. A substantial number of studies have been conducted to examine whether the suppressed effects of PAI-1 in type II epithelial cells would also be found in an animal experimental model of bleomycin-induced pulmonary fibrosis. According to this, in rats which were orally given tiplaxtinin, as compared with normal controls, the occurrence of pulmonary fibrosis was significantly suppressed. Besides, the concentration of TGF-β in the bronchoalveolar lavage fluid was also decreased. When the occurrence of pulmonary damage was induced by the treatment with bleomycin in rats, the expression of such pro-coagulant molecules such as PAI-1 or tissue factor was increased7. In addition, the expression of PAI-1 gene arises from reactive oxygen species that were increased by TGF-β and TNF-α21. Also in the lung of patients with idiopathic pulmonary fibrosis (IPF), as compared with normal healthy people, the concentration of PAI-1 was increased22. This implies that the fibrinolytic activity would also be decreased due to the PAI-1-induced suppression of tPA and uPA in the human body and this would play a key role in the pathogenesis of pulmonary fibrosis. As shown in the current results, however, PAI-1 promoted the EMT without suppressing PA. These results indicate that it might play a role in the pathophysiology of pulmonary fibrosis.
Limitations of the current study are as follows:
1) It was demonstrated that the PAI-1 was directly involved in the occurrence of CSE-induced EMT in type II epithelial cells. But no studies have been conducted to examine the PAI-1 receptors. There are no reports about the PAI-1 receptors. In the current experimental study, however, SMAD2 was phosphorylated by the CSE and then suppressed by PAI-1 siRNA. This implies that the TGF-β1 receptors might be shared.
2) It was not examined whether the EMT phenomenon would be suppressed by suppressing the expression of Snail mRNA with the use of siRNA whose target was Snail transcription factors in type II epithelial cells. In A549 cells stimulated by TGF-β1 and type II alveolar epithelial cells of mice, the expression of Snail mRNA was increased20,23 and Snail transcription factors played a key role in triggering the occurrence of TGF-β1-induced EMT and the renal fibrosis24. It can therefore be inferred that the PAI-1-induced EMT, seen in the current study, might undergo the signal transduction pathway which is very similar to the TGF-β1-induced EMT.
3) The expression of SNAI2 (Slug) was not observed. It has been known that SNAI2 as well as Snail plays a key role in patients with TGF-β1-induced EMT or idiopathic pulmonary fibrosis (IPF) and an animal experimental model of bleomycin-induced pulmonary fibrosis20. Also in the current study, SNAI2 might have played a similar role to Snail transcription factor.
To summarize, CSE triggers the occurrence of EMT where type II alveolar epithelial cells of rat origin are converted into the myofibroblasts. This might be associated with the phosphorylation of SMAD2 and ERK1/2 MAPK by the CSE-induced PAL-1 and the resulting intracellular migration of Snail transcription factors. Besides, there were such findings that the bleomycin-induced pulmonary fibrosis was alleviated following the oral administration of PAI-1 inhibitors in rats. This might also due to the inhibitory effects of EMT to some extents.








 XML Download
XML Download