

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Transient global amnesia (TGA) is a rare amnestic syndrome characterized by the sudden onset of selective antero- and retrograde amnesia with a time course of up to 24 hours. The clinical and neuropsychological spectrum of this syndrome has been characterized previously, and several etiological factors (e.g., migraine, focal ischemia, venous flow abnormalities, and epileptic phenomena) have been suggested to be involved in the pathophysiology, although no clear picture has emerged from these findings.1,2 The nature of the observed MRI changes and hence the pathophysiologic mechanisms underlying TGA remain unclear. Several pathogenic mechanisms have been suggested (e.g., migraine, cortical spreading depression, ischemia, venous flow abnormalities, and seizure activity), which might lead to impairment of cellular diffusion with subsequent diffusion-weighted imaging (DWI) changes.2 Several case studies using singlephoton-emission tomography have shown transient relative hypoperfusion in the temporal lobe during the acute phase of TGA.4-7 More recently, DWI has been used to detect high-signal abnormalities in the hippocampus, which provides strong evidence of a causative ischemic event.8 The signal abnormalities detected by DWI in TGA are predominantly spots 1 to 3 mm in diameter in the hippocampus.8 In one study, the lesions were mainly localized to the upper lateral portion of the hippocampus, which corresponds to the CA1 region9 and is known to be susceptible to hypoxia.10 However, in most of the other previous studies, DWI and/or T2-weighted imaging were not performed in the coronal plane, which made it difficult to localize the lesions precisely within the hippocampus. In the present study, we performed DWI and T2-weighted imaging in the coronal plane perpendicular to the hippocampus in patients with TGA. We hypothesized that TGA lesions would occur preferentially in the lateral portion of the hippocampus corresponding to CA1.
MATERIALS AND METHODS
1. Subjects
From October 2006 to June 2007, 20 consecutive patients with a clinical diagnosis of TGA at the emergency room of Seoul National University Bundang Hospital were enrolled for this study after excluding 1 patient without MRI within 1 day of symptom onset. The patients comprised 15 women and 5 men aged from 51 to 70 years (mean=59.2 years, Table 1). These patients underwent MRI including DWI from 5 to 23 hours (mean=13.8 hours) after symptom onset. The duration of the TGA attack was 8.2±2.9 hours (mean±SD). MRI was repeated 3 days later once the patients had recovered. All the patients fulfilled the following six TGA criteria proposed by Hodges and Warlow: (1) the attack must be witnessed and information must be available from a capable observer who has to be present for most of the attacks, (2) there must be a clear-cut anterograde amnesia during the attack, (3) clouding of consciousness and loss of personal identity must be absent and the cognitive impairment must be limited to amnesia, (4) there should be no accompanying focal neurological symptom during the attack and no significant neurological sign afterward, (5) epileptic features must be absent, and (6) the patient must not have had a recent head injury or active epilepsy.1
2. MRI Technique
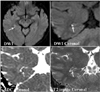
MRI was conducted using a 1.5-T unit (Intera, Philips Medical Systems, Best, The Netherlands) with a sensitivity-encoding (SENSE) head coil and a head/neck synergy coil. Patients initially underwent MRI using our stroke protocol, which consisted of DWI, T1- and T2-weighted, fluid-attenuated inversion-recovery, and conventional gradient-echo imaging in the transverse plane, three-dimensional TOF angiography of the intracranial region, and contrast-enhanced angiography of the neck region. Subsequent DWI and T2-weighted imaging in the coronal plane were performed perpendicularly to the long axis of the hippocampus with thinner slices. Coronal T2-weighted imaging was performed using the fast spin-echo technique with the following parameters: matrix=256×256 interpolated to 512×512, field of view=200-220 mm, slice thickness=3 mm, TR=4113 ms, TE=100 ms, echo-train length=24, SENSE factor=1, and number of acquisitions=3. DWI was performed in a transverse plane at 1.5 T initially within 24 hours after TGA onset (mean=12 hours, range=4-22 hours) and then after 3 days (mean=73 hours, range=71-75 hours) with four sequences: b=1000 (sec/mm2) and slice thickness=5 mm, b=1000 and slice thickness=3 mm, b=2000 and slice thickness=3 mm, and b=3000 and slice thickness=3 mm. These DWI sequences consisted of single-shot spin-echo echo planar imaging with the following parameters: matrix=128×128 interpolated to 256×256, field of view=220 mm, TR=5000-12,500 ms (5000 ms for b=1000, 9400 ms for b=2000, and 12,500 ms for b=3000), TE=60-75 ms, SENSE factor=2, and number of acquisitions=4. Detection rates of the lesion on DWI were compared based on the consensus of three neuroradiologists. If the lesion was positive on DWI, lesion conspicuity was graded as + or ++ (Fig. 1). All patients with high-signal abnormalities in the hippocampus on the initial DWI underwent subsequent DWI and T2-weighted imaging in the coronal plane to precisely localize the lesions.
3. Analysis
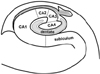
Locations of the lesions in the hippocampus (i.e., unilateral/bilateral and head/body/tail) were evaluated primarily using transverse DWI. The hippocampal lesions were localized more specifically on thin-section coronal DWI and T2-weighted images. Lesion locations were arbitrarily divided into lower medial, lateral, and upper medial regions (Fig. 2). According to the existing knowledge of the hippocampal anatomy,11 CA1 - the largest of the four hippocampal sectors (i.e., CA1-CA4) - occupies the lateral curvature of the hippocampus. Therefore, the lower medial, lateral, and upper medial regions correspond approximately to the subiculum, CA1, and remaining sectors (including CA2, CA3, CA4, and the dentate gyrus), respectively.
RESULTS
All patients had small punctuate (1-3 mm) DWI high-signal lesions in the hippocampus. The clinical data of the 20 patients are listed in Table 2. No abnormal findings other than hippocampal lesions were detected on DWI and other MRI modalities. In 17 patients the high-signal hippocampal abnormalities were unilaterally on DWI. One of these patients had two lesions in one hippocampus. Three of the 20 patients had lesions bilaterally in the hippocampus, 1 of whom had 3 bilateral lesions. A total of 25 lesions were identified: 5 in the hippocampal head, 19 in the body, and 1 in the tail. Six patients had unilateral lesions on the left, 11 patients had them on the right, and 3 patients had bilateral lesions. All lesions were found in the lateral portion of the hippocampus on transverse DWI (Fig. 3). All lesions were clearly demonstrated on both coronal DWI and T2-weighted images, and were localized to the lateral region of the hippocampus and corresponded to small areas of low signal intensity on apparent diffusion coefficient maps (Fig. 4).
DISCUSSION
One of the hypotheses for the etiology of TGA is transient cerebral ischemia. In contrast to the findings in TIA patients, the results of DWI studies in TGA patients are controversial. A variety of stressful events, including physical activity and emotional stress, are known to precede TGA in many cases. Lewis12 pointed out that TGA might be caused by venous congestion as a consequence of a retrograde transmission of high venous pressure after Valsalva-like activities. This hypothesis was confirmed by the finding that the incidence of a retrograde flow pattern in the internal jugular vein is elevated in TGA.13,14 Others have proposed a specific anxious personality trait in TGA patients that might induce vasoconstriction due to hyperventilation during emotional arousal.15 Therefore, temporary hypoperfusion either due to venous congestion or vasoconstriction due to hyperventilation could cause TGA. The increased vulnerability of the hippocampus and the venous drainage might help to explain why memory structures are the first to be involved.14 Regarding the thromboembolic hypothesis, some previous studies found no association between TGA and vascular risk factors,16 whereas others suggested an association with vascular risk factors. However, the thromboembolic hypothesis is still suggested for the etiology of TGA. Patients with TGA show a higher vascular risk profile, significantly more frequent carotid atherosclerosis, and an increased carotid intima-media thickness compared to patients without TGA patients.13 These findings might support an arterial embolic event, particularly in this subgroup of TGA patients.
Clinical and experimental data show that hippocampal CA1 neurons are critically involved in the process of memory consolidation by exhibiting a relay function in direct and polysynaptic intrahippocampal circuits, and lesions in this area are sufficient to produce clinically significant memory impairment. Pathophysiologically, neurons of the CA1 sector (Sommer sector) of the cornu ammonis are of particular interest since they show a selective vulnerability to cellular metabolic stress (e.g., during hypoxemia and ischemia) that leads to the glutamate- and calcium-induced and apoptosis mediated 'delayed neuronal death' of affected neurons 1-3 days after hypoxia. CA1 is known to be the sector most vulnerable to hypoxia, and is supplied by only one large ventral artery, whereas the other regions (i.e., CA2-CA4) are supplied by one large dorsal artery and several small arteries. Moreover, there are fewer microvessels in CA1 than in the other sectors.9 These anatomical differences could make CA1 more vulnerable to ischemia than the other sectors.
We suggest the following reasons for the low detection rate of DWI lesions during the first several hours after TGA onset. First, the lesion is too small and its DWI signal intensity is subtle at the hyperacute stage, making it susceptible to partial volume averaging due to the MRI sectioning thickness being larger than the lesion. We have also found in other investigations (separate from this study) that the lesion detection rate within 12 hours of TGA onset was higher for transverse DWI with a thinner section thickness (3 mm) and higher b value (2000 sec/mm2). Second, the pathogenetic mechanism of ischemia in TGA might differ from the more common thromboembolic ischemia. Delayed neuronal injury might occur, and the appearance of DWI lesions might also be delayed. In addition, a high signal intensity on DWI does not always indicate ischemia, as seen in other conditions such as multiple sclerosis and seizure.17 However, we speculate that a delayed appearance of a high DWI signal (which was too subtle to be detected initially) might result from the progressive T2 prolongation of the infarcted tissue. The high signal intensity of the infarctions on DWI after the first several hours results from a combination of diffusion restriction and T2 prolongation. Further developments of optimal DWI protocols for TGA that consider the section thickness, b value, and imaging timing might enable earlier and more accurate diagnosis, thereby aiding the documentation of heterogeneous DWI findings.
In conclusion, in this study lesions associated with TGA were localized mostly to the lateral portion of the hippocampus corresponding to CA1. This finding supports the ischemic etiology of TGA, but the underlying pathophysiologic mechanism requires further investigation.




 XML Download
XML Download