

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Negative myoclonus (NM) is a condition characterized by brief postural lapses of muscle tone, which can be demonstrated via electromyography (EMG) recordings.1,2 NM frequently occurs in association with metabolic encephalopathies. The EMG correlate of NM is a brief (<500 ms) silent period that is not preceded by any enhancement of EMG activity (i.e., myoclonus).3 Epileptic negative myoclonus (ENM) is defined as an interruption of tonic muscular activity that is time-locked to a spike on the electroencephalogram (EEG) without evidence of an antecedent myoclonus. ENM occurs in different epilepsy syndromes with a variety of etiologies; however, the exact origin of and mechanisms underlying the generation of ENM remain to be elucidated.1 Regarding the cortical areas that mediate NM, there is some evidence for the involvement of the perirolandic cortex, whereas studies on ENM have shown that this phenomenon may be associated with epileptic activity in the premotor cortex, including the supplementary motor area, or in the postcentral somatosensory cortex.3,4
There have been a few reports on ENM since its first description in 1981,4 but its association with strokes has not been described previously. Here we describe a patient who suffered an acute focal lesion as a result of a contralateral cerebral infarction; the stroke involved the right parietotemporal cortex, and the patient subsequently experienced the onset of unilateral partial motor epilepsy with NM, which is also referred to as an ENM.
CASE REPORT
A 62-year-old right-handed man was admitted to hospital with sudden-onset speech disturbance and left hemiparesis that first appeared 12 hours previously. His past medical record revealed an infarct in the territory of the left carotid artery that had occurred 2 years previously. He also had a 5-year history of hypertension, which was well controlled. His daily medications included aspirin at 100 mg, atenolol at 25 mg, and phenytoin at 300 mg.
The patient was alert and oriented, with fluent but mildly dysarthric speech, left central-type facial palsy, mild left hemiparesis (MRC, GIV+/V), numbness of the left extremities, and mild ataxia of the left upper limb; he also exhibited dyssynergia, hypermetria, and tremor during the finger-to-nose test with the left hand. Laboratory parameters, including a complete blood cell and platelet count, erythrocyte sedimentation rate, blood electrolytes, creatinine, liver enzymes, cholesterol, triglycerides, and prothrombin and partial thromboplastin time, were all normal. In addition, the serum level of phenytoin was 5.09 µg/ml.
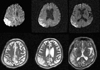
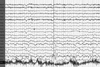
Brain magnetic resonance imaging (MRI) performed on his admission revealed high signal intensities in the right parietotemporal cortex in both diffusion- and T2-weighted imaging (Fig. 1). One day later, when the patient had regained almost complete strength in his left hand, he complained of an abnormal movement of his face and arm. These movements consisted of intermittent, involuntary, jerk-like movements of the left perioral area and arrhythmic losses of extensor muscle tone that developed upon instruction to maintain the wrist in an extended position. A waking, surface EEG revealed periodic epileptic discharges on the right parietotemporal area (mainly on leads T6 and P4). Polygraphic recordings, including the EEG and the surface EMG of the extensor wrist muscles, were performed to verify unilateral postural lapses of the left arm. With the patient's left upper limb at rest, the EEG epileptic discharges were not accompanied by any motor signs. EEG epileptic discharges in the right parietotemporal area occurred when the left upper limbs were outstretched, which were accompanied by 200- to 500-ms silent periods on surface EMG recording of the left wrist extensor muscles, and these provoked postural lapses of muscle tone (Fig. 2). The somatosensory-evoked potential recording (SEP) that was obtained following stimulation of the left median nerve showed normal latency and configuration findings, except for the decreased amplitude. These polygraphic recordings and SEP findings suggested the presence of ENM. The patient was treated daily with aspirin at 300 mg, dipyridamole at 400 mg, valproic acid at 2000 mg, and phenytoin at 200 mg. These adjustment of the patient's epileptic drugs resulted in an almost complete disappearance of both the ENM and the abnormal EEG epileptic discharges (Fig. 3).
DISCUSSION
ENM must be differentiated from nonepileptic positive myoclonus and asterixis, where the latter is not associated with the time-locked EEG correlates. Positive myoclonus can be ascertained easily by polygraphic recordings of the agonist and antagonist muscles.5 In our patient, simultaneous EEG and EMG polygraphic recordings showed postural lapses of muscle tone, which were time-locked with a sharp or spike wave over the contralateral parietotemporal cortex. Therefore, the most interesting feature in our patient was the peculiar focal inhibition of muscle tone related to EEG spikes that was not associated with myoclonic jerks.
The pathogenetic mechanism of ENM remains obscure, and that of the ENM evident in this patient also remains speculative. Ikeda et al. found that activation of the perirolandic cortex can produce pure NM, and they also provided evidence of the capability of the nonprimary motor areas - such as the premotor cortex and supplementary motor cortex - to generate NM.6 It was reported that using single electrical pulses to stimulate the premotor cortex, the primary motor cortex, the primary somatosensory cortex, and the supplementary motor area through stereo-EEG electrodes could induce pure silent periods on the EMG findings related to NM, not preceded by a motor evoked potential, which clinically appear as NM. These findings suggests that these cortical areas are involved in the genesis of NM.3,7 However, previous authors have pointed out that stimulation of the supplementary motor area induces only pure silent periods, regardless of the stimulus intensity, whereas the occurrence of pure silent periods following stimulation of the premotor cortex, primary motor cortex, and primary somatosensory area depend mainly on the stimulation intensity.3,7 This episodic dysfunction within the neural circuits that are normally concerned with the maintenance of sustained or tonic muscle contraction, so-called NM, may be released by focal lesions that are only in specific CNS areas or by a more generalized neurochemical imbalance (metabolic encephalopathies of various kinds, such as hepatic encephalopathy, renal and respiratory failure, and electrolyte imbalances).8,9 Several authors have also reported that high-frequency trains of electrical stimuli delivered directly to the negative motor area, including the premotor cortex, the primary motor cortex, the primary somatosensory cortex, and the supplementary motor area, can produce sustained negative motor responses, such as behavioral arrest, motor slowing, and inability to maintain a tonic contraction.3,10 Therefore, regarding the pathogenesis of ENM in our patient, we speculate that the lesion involving the parietotemporal cortex gives way to a strong intensity of abnormal electrical stimulation this intensity of electrical stimulation, which might be sufficient to effect pure silent periods, could spread from the postcentral cerebral cortex to the adjacent motor areaand thereby evoke pure silent periods, so-called ENM.
It is possible that the ENM in our patient was produced by either a metabolic imbalance or by neuropharmacologic agents, which caused a drug-induced encephalopathy.11-13 However, the relationships between the MRI and polygraphic evidence involving the postcentral somatosensory area and the response to anticonvulsant treatment, as well as the absence of other responsible etiologies, favors the argument of a cause -and-effect relationship, and this evidence weighs against the observations being coincidental. Despite a common cortical origin and some similarities in clinical semiology, to our knowledge, ENM and NM are two distinct entities. On the basis of several reports mentioned above and our own experience, ENM can be easily misdiagnosed. Thus, the appearance of ataxia, postural instability, or clumsiness in a patient with epileptic activity should suggest ENM when the symptoms cannot be ascribed to metabolic encephalopathy (including drug toxicity) or to progressive disease.
To the best of our knowledge, there are few reports on ENM-associated stroke in the medical literature, and there is still no consensus regarding the pathological mechanism underlying ENM. The case presented here suggests that a destructive lesion of the postcentral cerebral cortex and alteration of its outflow pathways to the neighboring motor area can lead to ENM.

 XML Download
XML Download