

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Epilepsy is a neurologic condition of diverse etiologies that affects about 1% of the global population.1 Approximately 325,000 children from 5 to 14 years of age have active epilepsy, and the majority of adults with epilepsy experience childhood onset of seizures.2 The median age of seizure onset is between 5 and 6 years.3 A single unprovoked seizure during childhood is common, occurring in 4 - 10% of children.4 Children in their first year of life, particularly, are at the highest risk for developing epilepsy.
Although the central nervous system (CNS) used to be considered an immunoprivileged system due to the presence of the blood-brain barrier (BBB), graft acceptance, a lack of conventional lymphatic drainage, and relatively low levels of monocytes and lymphocytes, it is becoming clear that immune and inflammatory reactions do occur in the CNS, either intrinsically from the brain itself or acquired from systemic circulation through a damaged BBB. While the role of inflammation in the pathophysiology of human epilepsy remains hypothetical, inflammatory and immune reactions in the brains do occur in human epilepsy patients and in experimental models of epilepsy.
Acute inflammatory reaction after seizures has long been suspected due to clinical observation of pleocytosis, without any evidence of infection, in the cerebrospinal fluid (CSF) and peripheral blood of patients with recent generalized convulsions. Steroids or adrenocorticotropic hormone (ACTH), with a suppressive effect on inflammation or immune reactions, have been used to effectively treat children with intractable epilepsy. Various common pediatric infectious or autoimmune diseases are often heralded by seizures at the onset or accompanied by seizures during the course of the illness.
INFLAMMATION IS IMPLICATED IN CHILDHOOD NEUROLOGICAL DISEASES ACCOMPANIED BY SEIZURES
Damaged BBB in CNS injury and delayed onset of epilepsy
The BBB consists of morphologically noninfestrated endothelial cells with interendothelial tight junctions, and its maintenance depends on normal functioning of pericytes, perivascular microglia, astrocytes, and the basal lamina. Under normal conditions, the BBB protects the CNS by regulating the entry of plasma-born substances and immune cells.5 Astrocytes are thought to act as important regulators of the balance between endothelial stability and permeability of the BBB. A high transendothelial barrier can be reintroduced in human or bovine endothelial cell monolayers cultured in astrocyte-conditioned media, suggesting that astrocyte-derived soluble factors may contribute BBB characteristics to endothelial cells.6 Transient changes have been demonstrated in the physiology and structures of the BBB in various CNS injuries such as status epilepticus, infections, and traumatic and ischemic events.5 An impaired BBB and inflammatory state are common features of neurological diseases associated with the late onset of epilepsy.7 Proinflammatory cytokines are elevated in experimental animal brains after ischemia8 and in the CSF from stroke9 and epilepsy10 patients. Cytokine release causes subsequent up-regulation of endothelial and neutrophil adhesion molecules in human cerebrovascular endothelial cells during hypoxic injury,11 leading to transmigration of leukocytes across the endothelium and the BBB. Leukocyte recruitment may trigger signal transduction cascades, resulting in tight junction disorganization and BBB breakdown. Although the mechanism of delayed onset of epilepsy remains unclear, available data suggest that inflammation and breakdown of the BBB are necessary components of epileptogenesis following brain injury. Further work is needed to determine whether BBB breakdown is a pre-requisite for future development of epilepsy and to elucidate the potential for prophylactic treatment during the latent period following an injury to prevent epilepsy.
Childhood noninfectious neurological diseases associated with the late onset of epilepsy
Neonatal and childhood stroke
The incidence of arterial ischemic stroke and cerebral sinovenous thrombosis has increased to 2 to 6 per 100,000 children a year during the past 10 years.12 Even in neonates, stroke affects as many as 1 in every 4000 live births.13 Common risk factors include congenital heart disease and sickle cell disease. Stroke in infancy and childhood adversely affects development; neurological deficits occur in 60% of children and 10 - 25% will experience recurrent stroke.14 Treatment is empirically provided with antithrombotic drugs.
In perinatal stroke, neonatal seizures are the most common clinical symptom. Neonatal seizures in a setting of arterial stroke are mostly focal and may occur in the absence of other signs of neonatal encephalopathy. Of infants with ischemic cerebral infarcts proven in autopsy studies, 25 - 40% are diagnosed with neonatal seizures.15 Some infants with a normal neurological examination in the neonatal period may be diagnosed in later months with asymmetry of reach and grasp, failure to reach developmental milestones, or post-neonatal seizures.16 Neonatal seizures and abnormal neurologic examination at discharge are two risk factors for later neuro-developmental disabilities and chronic epilepsy.17 Childhood epilepsy is a frequent resulting morbidity in perinatal stroke. In one cohort study of 64 perinatal stroke patients, 75% of children presented with neonatal seizures and 67% developed epilepsy after 6 months of age.18 The median age at onset of delayed epilepsy was 16 months.
In childhood stroke, seizures commonly occur acutely and may be the presenting symptom of stroke. A study of 73 children 17 years old and younger with acute hemiplegia from stroke found that 50% of patients had at least one seizure and 29% had recurrent seizures.19 Time to onset of delayed seizures and development of epilepsy ranged from 4 months to more than 10 years in 42 children after unilateral hemispheric stroke.20
Proinflammatory cytokines IL-1β and IL-6 increase in CSF from stroke patients within the first 24 h after the beginning of symptoms.21 Systemic injections of progesterone, a neurosteroid, improve cognitive recovery after stroke and decrease molecular indicators of neuronal damage in rats,22 suggesting immunosuppression as a possible treatment strategy to prevent late sequelae.
Autoimmune diseases and epilepsy
a. Systemic Lupus Erythematosus (SLE)
SLE is the most common rheumatic disease in children.23 The major organ systems involved in childhood SLE are similar to adult SLE, but the frequency of multiple organ involvement and severity of the disease are greater in children than adults.23 General clinical features include broad variations of rash, arthritis, constitutional symptoms, renal disease, and cardiovascular, pulmonary, and neuropsychiatric involvement. The prevalence of epilepsy in SLE patients is 10 - 20%, 8 times higher than in the general population. Notably, seizures can precede the diagnosis of SLE, and 5 - 10% of patients experience seizures several years before the clinical onset of SLE.24 This finding suggests that long-term treatment with antiepileptic drugs may have precipitated SLE, or alternatively, that epilepsy and SLE are both manifestations of a genetically determined predisposition to altered immunity and inflammatory reaction. Epilepsy in SLE is significantly associated with anti-phospholipid antibodies, and the presence of anti-phospholipid antibodies highly correlates with abnormal MRI findings of the brain.24 Patients with anti-phospholipid antibodies are at risk of thromboembolic manifestations, intrauterine fetal loss, and thrombocytopenia, a combination termed anti-phospholipid syndrome. In addition, anti-cardiolipin antibodies are found in 30 - 60% of SLE patients, and epilepsy is 3 times more frequent in patients with anti-cardiolipin antibodies than those without.25 These findings raise the possibility that autoantibodies may trigger seizures and contribute to epileptogenesis.
b. Hashimoto thyroiditis
Hashimoto thyroiditis is the most common thyroiditis in children and adults, affecting 1.2% of school-aged children.26 Hashimoto encephalopathy is a syndrome associated with high antithyroid antibody titer that can either begin abruptly, in the form of seizures or agitation, or develop gradually, in a relapsing-remitting manner manifested as cognitive deterioration and psychiatric illness. The occurrence of Hashimoto encephalopathy is unrelated to the patient's thyroid function status, and most patients respond dramatically to corticosteroid therapy.26 The therapeutic efficacy of immunomodulation suggests a causative role of activated immunity and autoantibodies for the neurological symptoms in Hashimoto thyroiditis, including seizures and epilepsy.
c. Behçet's disease
Behçet's disease is a chronic systemic inflammatory disorder of unknown etiology defined by the classical triad of recurrent oral aphthous, genital ulcers, and inflammatory eye disease. The prevalence of CNS disease, often associated with significant morbidity, varies between 2.9% and 44%, with male predominance.27 In 22 patients with neuro-Behçet's disease, 27% suffered either single or recurrent seizures.28 CSF pleocytosis occurred in 50% of patients, and most showed improvement after immunosuppressant therapy.
d. Rasmussen's encephalitis
Rasmussen's encephalitis is a prototype of inflammatory epilepsy. The autoimmune nature of this condition was suspected after the discovery of autoantibodies against glutamate receptor, GluR3, one of the AMPA (α-3-hydroxy-5-methyl-4-isoxazolepropionic acid) subunits. Subsequently, anti-GluR3 antibodies have been detected in two other epilepsy syndromes, early-onset noninflammatory focal epilepsy and catastrophic infantile epilepsy.29 Anti-cardiolipin antibodies, found in SLE patients, are also found in epilepsy patients, both adults30 and children.31,32 Furthermore, increased levels of anti-nuclear antibodies have been reported in epilepsy patients.30 Anti-B2-glycoprotein I antibodies, specific for thrombosis-mediated events, have been demonstrated in SLE patients with epilepsy33 as well as other epilepsy patients.30 Long-term immunotherapy such as intravenous γ-globulin and corticosteroids may be beneficial for autoantibody-positive epilepsy patients.
Role of systemic and brain inflammation in pathogenesis of febrile seizures
Febrile seizures are the most common cause of seizures in children, affecting 2 to 5% of children.34 The threshold to febrile seizures is dependent on the height of the body temperature, but the threshold varies with individuals and according to age and maturation.35 A genetic susceptibility to inflammation may influence the threshold convulsive temperature. Seventeen to 30% of febrile seizure patients have a family history of febrile seizures.35 A biallelic polymorphism in the promoter region of IL-1β at the -511 position that can increase IL-1β production occurs more frequently in patients with prolonged febrile convulsions.36,37 In experimental animals, intraventricular injection of IL-1β reduces the seizure threshold in 14-day old mice subjected to hyperthermia, while IL-1receptor knock-out mice have higher seizure thresholds, supporting the role of proinflammatory cytokines in triggering febrile seizures.38
Viruses as being increasingly implicated as causative agents of febrile seizures. Neurotropic viruses, such as the herpesviruses and influenza A, are commonly associated with febrile seizures in the United States and Asia.39,40 Fever induced by viral infection is regulated by components of the immune response, particularly proinflammatory cytokines. Proinflammatory cytokines are higher in influenza-associated febrile seizures, further suggesting a causative role for cytokines in the pathogenesis of febrile seizures. Common pathogens and causes associated with febrile seizures are detailed below.
Human herpesvirus-6 (HHV-6) infection
HHV-6 causes exanthema subitum, an acute febrile illness affecting more than 90% of children before 2 years of age, the period of greatest susceptibility to febrile seizures. The incidence of convulsions among exanthema subitum varies from 2 to 50%.35,39,41,42 The incidence of primary HHV-6 infection is similar between first simple febrile seizure patients and age-matched controls.43 HHV-6 DNA, however, is detected more frequently in CSF of patients with three or more seizures than those with a single febrile seizure.44 This finding suggests that HHV-6 may invade the brain during the acute viremic phase of exanthema subitum and then reactivate when triggered and provoke recurrent febrile seizures.
Influenza virus
Influenza virus A is a frequent cause of febrile seizures in Japan45 and China.40 During 1997-1998, influenza A accounted for 35 - 44% of hospital admissions for febrile seizures in Hong Kong.40 Influenza-associated febrile seizures are often prolonged and complex, independent of the severity of the viral infection.45 IFN-α and IL-6 are significantly higher in patients with febrile seizures compared to those without.46 This finding suggests that influenza-associated febrile seizures may be the result of systemic immune responses, and that IFN-α and IL-6 are involved in the pathogenesis of febrile seizures (in influenza, at least).
Vaccines
Vaccines, including measles-mumps-rubella (MMR) and diphtheria-tetanus-pertussis (DTP), are associated with an increased risk of febrile seizures in the first three years of life.47 MMR-associated seizures often occur at 7 to 14 days after vaccination, while DTP-associated seizures typically occur on the day of vaccination.48 Children experiencing febrile seizures associated with vaccination have greater rates of family history of febrile seizures and a high risk for recurrent febrile seizures47,49 Although it remains unclear whether vaccination itself represents a risk, or if fever after vaccination is necessary for seizures, immunization-provoked seizures may imply a role for systemic inflammation and genetic susceptibility in the induction of febrile seizures after vaccination.
Benign afebrile seizures in acute gastroenteritis
Benign seizures associated with diarrheal illnesses are afebrile generalized tonic-clonic convulsions that occur between the 1st and the 5th sick day of viral gastroenteritis in previously healthy young children 6 months to 3 years of age.50 Rotavirus antigen has been detected in the majority of patients with this condition and rotaviral RNA has been found in patients' CSF.51 Rotavirus enterocolitis is the most common enterocolitis among young children under 2 years of age. Rotavirus has tropism for astrocytes in astrocytoma cell lines,52 and inoculation of rotavirus strain 2 into the brains of live monkeys can induce transient symptoms reminiscent of typical encephalitis (typical infection of the CNS).53 Since only 6.4% of children with rotavirus gastroenteritis present with afebrile benign seizures,54 it may be that only a few specific strains of rotavirus can infect both the CNS and gastrointestinal system or that host susceptibility factors make a subset of children more prone either to CNS invasion by rotavirus or to specific immune responses to rotavirus.
Medically intractable childhood epilepsy improved by immunotherapy
Nearly 30% of epilepsy patients are refractory to conventional anti-epileptic drugs, and many alternative treatments have been tried to control epilepsy.55 Immunotherapy, such as corticosteroids and ACTH, has been used to treat epilepsy since ACTH was first reported to have beneficial effects in the treatment of infantile spasms in 1950.56 The mechanism behind the anticonvulsant action of corticosteroids or ACTH remains elusive. Possibilities include (1) stimulation of glucocorticoid synthesis that interacts with CNS steroid receptors, which then influences voltage-dependent calcium channels; (2) stimulation of neurosteroid synthesis in glia and neurons that modulate GABAA receptors; (3) down-regulation of corticotrophin releasing hormone (CRH) that has proconvulsant activity in the immature brain; or (4) immunomodulation.57-60
Rasmussen's encephalitis
Rasmussen's encephalitis is a severe, progressive focal epilepsy of unknown origin that leads to asymmetric cortical atrophy and deterioration of motor and cognitive function.61 The current histopathological criteria of Rasmussen's encephalitis include the presence of T-cell-dominated inflammation, microglial activation and formation of microglial nodules, neuronal loss, and astrocytic activation. Recently, astrocytic apoptosis and subsequent loss have been demonstrated as a specific feature of Rasmussen's encephalitis.62 A specific attack by cytotoxic T lymphocytes may be one possible mechanism responsible for astrocytic degeneration in Rasmussen's encephalitis.62
Antibodies to GluR3 have been found in the serum of some Rasmussen's encephalitis patients, and repeated plasma exchanges can reduce serum titers of GluR3 antibodies, decrease seizure frequency, and improve neurologic function.63 Immunization of animals with GluR3 induces a disorder resembling the human disease.63 At the same time, a recent study reported that anti-GluR3 antibodies are not specific for Rasmussen's encephalitis. High antibody titers characterize a subgroup of non-Rasmussen's encephalitis patients with "catastrophic" epilepsy and appear to be a marker for intractable epilepsy not limited to Rasmussen's encephalitis. Anti-GluR3B peptide antibodies are significantly associated with seizure frequency.29
There is no effective medical treatment for Rasmussen's encephalitis, except perhaps steroids, which can be useful when given early in the course of the disease.64 A long term follow-up of 11 Rasmussen's encephalitis patients who received steroids showed that 45% of patients had significant improvement of motor function and reduction of seizure frequency with disappearance of epilepsia partialis continua, while 55% patients had no benefit from steroid therapy and ultimately underwent hemispherotomy. Two initial responders to steroid treatment experienced progressive recurrence of seizures one to four years after the discontinuation of steroids and received a hemispherotomy.64
Infantile spasms
Infantile spasms are a unique, age-specific epilepsy of early infancy. The electroencephalogram (EEG) characteristically shows hypsarrhythmia-disorganized, chaotic, high voltage polymorphic delta and theta rhythms with superimposed multifocal spikes and wave discharges. The onset of spasms is frequently associated with neuro-developmental regression. The incidence varies from 0.25 to 0.60 per 1,000 live births,65 and the prevalence rate is 0.15 to 0.2 per 1,000 children age 10 or younger.66 Infantile spasms develop in infants with a variety of CNS pathologies, including structural abnormalities, prenatal and postnatal infection, stroke, trauma, and chromosomal anomalies.67 This disease entity of uniform semiology with diverse causes suggests that infantile spasms are an age-specific but cause-nonspecific final common response of the brain to insults.68,69
ACTH is a well-known effective treatment for infantile spasms that not only results in seizure control, but also improves both behavior and background EEG.70 Meta-analysis reveals that ACTH is probably effective for short-term treatment of infantile spasms and leads to resolution of hypsarrhythmia.71 Time to response is usually two weeks. Oral steroids can render 30 to 40% of patients seizure-free.72,73 Further, early use of steroids is more effective; patients treated within one month of spasm onset had a better outcome than those treated after more than one month.74 A study of 18 children with Down's syndrome and infantile spasms showed that spasm control was easier, subsequent seizures were less persistent, developmental quotients were higher, and a score of autistic features was lower in children treated early.75
Lennox-Gastaut syndrome
Lennox-Gastaut syndrome (LGS) is diagnosed in children who have a minimum of two seizure types (usually atonic or astatic (drop) seizures, nocturnal tonic seizures, and atypical absence seizures), mental retardation, and bursts of generalized slow spike-waves on EEG.76 LGS is one of the epilepsy syndromes most resistant to the available antiepileptic medications. Corticosteroids and ACTH may be effective in treating children with LGS.55 When 10 children with LGS and intractable seizures were treated with prednisolone for 12 weeks, 7 achieved seizure freedom and the other 3 experienced seizure reduction.77 The patients' clinical improvement was generally reflected in an improvement in their EEGs. Another study of 45 children with LGS who received ACTH for 2 to 8 weeks found that while 51% of patients became seizure free for over 10 days, 78% of these children later relapsed.78
Landau-Kleffner syndrome (LKS)
Landau-Kleffner syndrome is an acquired epileptic aphasia presenting as progressive loss of speech in a previously well child with an abnormal and usually continuously epileptic EEGs during slow wave sleep with or without apparent seizures.79 There are reports that corticosteroids improved both clinical features of the syndrome as well as the EEG, especially when instituted early.80,81
Epilepsy with continuous spike wave discharges (CSWSS)
Continuous spike-waves during slow sleep syndrome (CSWSS) is a rare, sporadic childhood epilepsy characterized by the presence of spike-waves during at least 85% of slow sleep and, clinically, by the existence of neuropsychological and behavioral disorders.82 LKS and CSWSS share some common features, including first appearance in childhood and mild epilepsy associated with severe neuropsychological disturbances. There are several case reports that CSWSS also responds to treatment with corticosteroids or ACTH, not only in terms of seizures and EEG, but also in language skills.83
Myoclonic or myoclonic-astatic seizures and other refractory epilepsies
Myoclonic or myoclonic-astatic seizures have also been treated with corticosteroids, possibly because of the frequent daily seizures, acute encephalopathic presentation, medical intractability, and tendency of the seizures to result in physical injury. In a study of myoclonic epilepsy, 34 children out of 64 were treated with prednisone and the remaining 30 children with ACTH.84 Seizures were effectively controlled in 73% of children treated with ACTH, but in none treated with prednisone.84 In another study of nine patients with myoclonic epilepsy treated with steroids, five became seizure free, two showed a reduction, and the remaining two exhibited no change.77 In contrast, only a transient improvement was reported in 5 of 84 patients with myoclonic absence epilepsy treated with ACTH.85 In a study of children with absence epilepsy, seven were treated with steroids. All improved and five became seizure free.77 In 32 children with intractable epilepsy (infantile spasms excluded) treated with ACTH, 25% of patients became seizure-free and 47% showed significant reductions of seizures.86
SEIZURES CAUSE ACUTE AND CHRONIC BRAIN INFLAMMATORY REACTIONS - Evidence from experimental models of epilepsy
In many animal models of epilepsy, acute seizures cause glial activation and increased expression of transcription factors and cytokines that coordinate inflammatory responses.87,88 After status epilepticus, members of the Toll-like receptor (TLR) family are significantly up-regulated in the microglia, leading to transcriptional activation of cytokines, chemokines, MHC class I and II, and costimulatory molecules.89 The activated glia and elevated cytokines, in turn, contribute to seizure-related hippocampal pathology, such as neuronal death, neuronal birth, reactive gliosis, and mossy fiber sprouting.90-93 Accumulating experimental data also suggest that seizure-induced glial activation and up-regulation of pro-inflammatory cytokines can lead to neuronal excitability and neuronal injury either directly, by interacting with glutamatergic neurotransmission, or indirectly, by activating gene transcription.
Microglial activation
Microglia are myeloid lineage cells that comprise approximately 12% of the brain. Resting microglia with a ramified morphology are responsible for immune surveillance94 and are activated at a very early stage in response to injury or immunological stimuli with transformation to an amoeboid shape.95 Activated microglia up-regulate the expression of surface molecules, such as complement receptors and major histocompatibility complex (MHC) molecules, and release a variety of pro-inflammatory and cytotoxic soluble factors.96 MHC class-I and -II, IL-1, IL-2, IL-6, TGF-β1, the complement components and their receptors, M-CSF, and GM-CSF are all molecules considered to be signals in the activation process.95
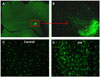
Widespread microglial activation accompanied by neuronal injury occurs after acute seizures.88,97,98 As a rapid response, activation of microglia may be responsible for neurodegeneration rather than a consequence of neurodegeneration. Within four hours after KA-induced status epilepticus, glial activation and cytokine expression are found in the hippocampus (Fig. 1).99 Neuronal injury is detected 12 - 24 hours following status, many hours after cytokines are induced in the glia.99 Status epilepticus, prolonged seizures over 30 minutes, can cause neuronal death100 through glutamate-mediated excitotoxicity, necrosis, and activation of apoptosis.101 One to three days after status epilepticus, both neuronal and astrocytic death are observed in the dentate hilus within the hippocampus.102,103 Injured neurons and glia and their fragmented DNA are rapidly cleared by activated microglia.93 The impaired neurogenesis in inflammation can be effectively restored by systemic administration of the tetracycline derivative minocycline, a specific inhibitor of microglia activation.104 These findings suggest that microglial activation associated with inflammation induces neuronal injury and suppresses neurogenesis. The occurrence of spontaneous seizures has been correlated with the extent of glial activation, as well as astrocyte and neuron degeneration in the hippocampus, and blockade of neuronal death failed to prevent epileptogenesis.105,106
Cytokines released by microglia influence astrocytic function and proliferation. Increased levels of IFN-γ, IL-1, IL-2, IL-6, TNF-α, and M-CSF are associated with astrogliosis. IL-1Ra, an IL-1 receptor antagonist, is sufficient to prevent astroglial proliferation, suggesting a pivotal role of IL-1 in astrocyte activation.107 In addition, these cytokines may modulate glutamate homeostasis by regulating glutamate receptors and transporters in astrocytes.108,109 Impaired handling of extracellular glutamate by gliotic astrocytes may lead to neuroexcitability and excitotoxic neuronal damages resulting from excessive glutamate levels.110 These findings suggest that activation of microglia and the resultant increase in cytokines may influence epileptogenesis by altering glutamatergic transmission indirectly through modulating astrocytes.
Astrogliosis
For over 100 years, astroglial proliferation has been observed as a pathognomonic finding in surgically resected hippocampi in patients with intractable mesial temporal lobe epilepsy. Once regarded as an inert scar and consequence of the healing process after neuronal degeneration,111 recent studies112,113 suggest that reactive astrogliosis and modified astroglial function may have an important role in the generation and spread of seizure activity.
Astrocytes are a critical component of the BBB and have many important roles in glutamate and potassium uptake and the production of growth factors, cytokines, and extracellular matrix proteins.104 In response to immunologic challenges or brain injuries, astrocytes proliferate and become hypertrophic and fibrillary. Direct stimulation of astrocytes by photolysis of caged calcium and glutamate release in acute seizure models can induce a paroxysmal depolarization shift (PDS) - abnormal prolonged depolarization observed during interictal activity.112 Cultured astrocytes isolated from the seizure focus of human epileptic tissue possess depolarized resting membrane potentials and are able to generate action potentiallike responses upon injection of current.113 Astrocytes are initially activated by excessive neuronal activity, a potent trigger of astrocyte Ca2+ signaling. However, once activated, neuronal firing is no longer needed for continued astroglial activation. The findings described above suggest that activated astrocytes in the sclerotic hippocampus are not mere bystanders, but active players in epileptogenesis.
Within 24 to 48 hours after seizure induction, activation of GFAP-positive astrocytes is noted throughout the dentate gyrus and hippocampal subfields.107,115 This reactive astrogliosis persists chronically over three to four months.105,114,115 Newly generated astrocytes display distinct changes in glial membrane channels and receptors to promote neuronal hyperexcitability and seizure generation. These changes include GluR1 receptors with elevated flip-to-flop ratios,116 impaired uptake of potassium ion due to inwardly rectifying K+ channels,117 over-expression of adenosine kinase,118 and down-regulation of glutamine synthase, glutamate dehydrogenase, and glial GABA transporter.102
Increased inflammation-related gene expression in the brain after status epilepticus
Gene expression of classical inflammatory mediators - the complement pathway, cytokines, and glial markers - is significantly higher in the hippocampus of both juvenile and adult rats after KA-induced seizures, based on microarray analysis in our laboratory (Fig. 2).119 Inflammation-related genes were the single most abundant functional group significantly up-regulated acutely after seizure, and significant elevations persisted even 10 days after SE. In juvenile rats, the acute inflammatory response appears to be part of the homeostatic response, because there is no cell death and no recurrent seizures. Experiencing seizures and an acute inflammatory reaction are not without consequence, however, and the rats become permanently more seizure-susceptible. On the other hand, the inflammatory response in adult rats appears to be chronic, excessive, and persistent, and is accompanied by seizure-induced cell death and subsequent development of spontaneous recurrent seizures only in older animals. Such age-dependent increases are reflected as microglial activation in the hippocampi.98,99
Pro-inflammatory cytokines and neuronal hyperexcitability
Experimentally induced seizures in rodents trigger a prompt inflammatory response in brain areas recruited in the onset and propagation of epileptic activity.90 Direct intra-cerebral injection of a cytokine worsens seizure activity,120,121 and cytokine receptor antibodies, such as IL-1-receptor antagonist, show powerful anticonvulsant activity.122
The mechanisms by which cytokines lead to neuronal excitability have been explored in several studies focusing on ictogenic and neurotoxic properties of IL-1β mediated by IL-1 receptor (IL-1RI).120,123 IL-1RI colocalizes with NMDA receptors on hippocampal pyramidal neurons, and IL-1RI-mediated modulation of glutamatergic transmission may contribute to excitotoxicity and spontaneous seizures.124 IL-1β binding to its receptor increases NMDA receptor-mediated Ca2+ influx and surface expression of AMPA receptors.125,126 IL-1β acts on astrocytes to inhibit reuptake of glutamate127 and increase glutamate release via TNF-α production,128 both resulting in elevated extracellular glutamate levels and hyperexcitability. Furthermore, IL-1β can stimulate IL-6 release.108 Transgenic mice over-expressing astrocyte IL-6 show markedly increased astrogliosis and microgliosis, a loss of inhibitory interneurons, and are exquisitely and selectively sensitive to glutamatergic agonists.129
Cytokines are involved in the death of neurons. Direct intraventricular IL-1β injection concurrent with brain insult increases injury-induced cell death and brain edema.130,131 Inhibition of IL-1 signaling by IL-1 receptor antagonist injection just prior to chemoconvulsant administration significantly attenuates subsequent hippocampal cell loss, implicating endogenous IL-1 in seizure-associated cell death.132
Cytokines can influence the activation of astrocytes and microglia. Astrogliosis can be induced in healthy animals by the injection of cytokines such as IL-1,133 and injury-induced microglial activation is suppressed in TNF receptor-knockout mice.134 In turn, reactive microglia and astrocytes themselves provide a rich source of cytokines after injury or insult, especially IL-6, TGF-β, LIF and IL-1.135
Lessons from transgenic mice
Transgenic or knock-out mice technology provides a powerful tool for studying genetic control of synaptic excitability and epilepsy. It is relatively easy to insert, delete, or mutate a gene of interest or to study the function of spontaneous mutation of genes causing epilepsy. A chronic brain inflammatory state has been induced in transgenic mice overexpressing specific cytokines to study the functional outcome of inflammatory mediators in the brain.
Transgenic mice with chronic IL-6 production from astrocytes using the transcriptional control of the GFAP promoter display astrogliosis, microgliosis, impaired development of the BBB, and increased levels of acute-phase proteins and proinflammatory cytokines.136,137 These mice develop progressive neurodegeneration in the hippocampus and cerebellum, which in turn contributes to a clinical picture of ataxia, tremor, progressive learning deficits, hippocampal excitability, and spontaneous seizures.137-139 GFAP-IL-6 mice show markedly enhanced sensitivity to glutamatergic-induced seizures and lethality, and this may relate to a compromise of inhibitory paralbumin- or GABA interneurons.129
In IL-6 null mice, reactive astrogliosis and microgliosis are significantly lower after KA-induced seizure, while morphological hippocampal damage, oxidative stress, and apoptotic neuronal death are greater. IL-6 deficiency impairs the inflammatory response after KA-induced seizures and increases neuronal injury.140 IL-6 is a major inducer of metallothionein I and II (MT-I+II), antioxidants and neuroregenerative factors in the CNS.. Decreased MT-I+II levels in IL-6 null mice may contribute to greater oxidative stress and cell death. These findings support the beneficial and protective role of the physiological inflammatory reaction and caution against potential exacerbation of neuronal injury by complete obliteration or blockage of the inflammatory cascade.
Transgenic mice deficient in IL-1β receptor 1 are resistant to experimental febrile seizure. Moreover, intracerebral injection of high dose IL-1β, sufficient to generate limbic seizures in wild type mice, fails to cause seizures in these mice.38 This finding suggests that IL-1β signaling contributes critically to fever-induced hyperexcitability underlying febrile seizures and may provide a link between hyperthermia and genetic susceptibility to seizure.
Epileptic EL mice, a natural model of human multifactorial idiopathic epilepsy and complex partial seizures, experience about 25 - 30 complex partial seizures with secondary generalization during routine weekly cage changing. Prior to the onset of recurrent seizure activity, the number of GFAP-positive astrocytes is similar in EL and non-epileptic mice at young ages.141 Recurrent seizure activity in EL mice produces a unique type of gliosis where microglial activation is diffuse and widespread throughout the cortical surface, including the hippocampus, while astrocyte activation is specifically localized to the hippocampus accompanied by down-regulation of glial glutamate transporters, but without obvious hippocampal neuronal loss or mossy fiber sprouting.142,143 These findings suggest a potential contributory role of glial abnormalities in the recurrent seizure activity of human multifactorial idiopathic epilepsy.
INFLAMMATORY REACTIONS IN CHRONIC EPILEPSY PATIENTS -Evidence from human epilepsy
Increased expression of proinflammatory molecules has been demonstrated in brain tissue from patients who received surgery for drug resistant epilepsies.99,144-147 Of note, inflammatory reactions occur not only in classic inflammatory epilepsy such as Rasmussen's encephalitis, but also in common intractable epilepsy that does not typically invoke an inflammatory pathophysiology, such as temporal lobe epilepsy or tuberous sclerosis. Brain inflammation may be a common factor contributing or predisposing to the occurrence of seizures and cell death in various forms of epilepsy of different etiologies.148
Blood and CSF from epilepsy patients
Acute inflammatory reaction after seizures has long been suspected due to clinical observations. Mean peripheral blood and CSF-leukocyte counts as well as C-reactive protein, an acute phase reactant, are significantly elevated in patients who present with new onset generalized convulsions without any clinical evidence of CNS or systemic infection.10 Involvement of the "cytokine network" has also been implicated in febrile seizures. Significantly higher plasma IL-6 levels as well as a higher ratio of endogenous IL-1 receptor antagonist to IL-1β are found in children with febrile seizures compared to febrile children without seizures36 and in epilepsy patients with recent tonic-clonic convulsions.149-151
Cytokine gene polymorphism has been linked to epilepsy susceptibility. The increased frequency of a biallelic polymorphism in the promoter region of IL-1β at the -511 position, which is suggested to have effects of higher IL-1β production, has been reported in patients with temporal lobe epilepsy with hippocampal sclerosis and prolonged febrile convulsions.37 Sporadic simple febrile seizure patients exhibit significantly higher frequencies of IL1β -31C/-511T alleles and homozygotes than controls.152 These results suggest that genetic susceptibility to inflammation may contribute to epileptogenesis.
Epileptogenic brains of adult epilepsy patients
Active inflammation has been detected not only in prototypical inflammatory epilepsy such as Rasmussen's encephalitis or limbic encephalitis, but also in pharmacoresistant epilepsy of diverse etiologies. Microglial activation and proliferation havebeen demonstrated in surgically resected epileptogenic lesions from adult patients with chronic intractable epilepsy. In human mesial temporal lobe epilepsy, activated microglia are increased over ten fold in the sclerotic hippocampi.153 An abundant population of activated microglial cells are found in glioneuronal lesions associated with chronic epileptic activity,154 in focal cortical dysplasia155 and in cortical tubers of the tuberous sclerosis complex.146 The duration of epilepsy and frequency of seizures prior to surgical resection significantly correlate with the density of activated microglia in focal cortical dysplasia,155 highly epileptogenic gangliogliomas, and dysembryoplastic neuroepithelial tumors (DNET).154 Activation of microglia appears to be an important feature of chronic intractable epilepsy, and microglial proliferation may be functionally related to epileptogenic brain lesions of diverse etiology. In addition, pro-inflammatory cytokines, such as IL-1b and its signaling receptor IL-1R1, and NF-kB are highly expressed by neurons and glia in temporal lobe epilepsy,145 focal cortical dysplasia,155 glioneuronal tumors,154 and tuberous sclerosis complex.146 These results strongly support the involvement of inflammatory and immune reactions in the pathogenesis of human CNS disorders associated with epilepsy.
Epileptogenic brains of children with epilepsy
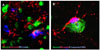
In our laboratory, we quantified cell death, astrocyte proliferation, microglial activation and cytokine release in brain cortical tissue from 13 children who underwent epilepsy surgery. Patients had intractable epilepsies due to focal cortical dysplasia (6), encephalomalacia (5), Rasmussen's encephalitis (1), or mesial temporal lobe epilepsy (1). Five autopsy patients with no history of seizures or neurological diseases were used as controls. We found marked glial activation and neuroinflammation in epileptogenic cortices from children with intractable epilepsy (Fig. 3). The majority of our patients had mental retardation. Numerous fibrillary astrocytes covered the entire cortex and converged on to blood vessels, neurons, and microglia. Large numbers of neurons and astrocytes displayed DNA fragmentation and the magnitude significantly correlated with seizure frequency. Panlaminar astrocytosis, diffuse microglial activation, and release of proinflammatory cytokines, especially IL-1β, IL-8, IL-12p70 and MIP-1β, were present in the epileptogenic cortices, consistent with chronic neuroinflammatory responses. IL-6 and MCP-1 were significantly higher in patients with a family history of epilepsy, suggesting links between genetic susceptibility to inflammation and epilepsy. Our results also suggest that active neuroinflammation occurs in pediatric epilepsy and may play a common pathogenic role in childhood epilepsies of diverse etiologies.
CONCLUSION
Accumulating evidence suggests that inflammatory and immune reactions may play an important role in neuronal excitability and epileptogenesis. Chronic brain inflammation may also contribute to intractability of seizures and comorbidity in chronic epilepsy patients. No effective treatments currently exist to protect the brain from seizure-induced cell death and prevent future development of chronic epilepsy. Modulation of inflammatory reactions in the brain and targeting of inflammatory mediators may be effective therapeutic strategies to prevent or limit epileptogenesis in the vulnerable nervous system. Anti-inflammatory therapy may be particularly helpful when given during the latency period shortly after the initial neurologic insult, but prior to the onset of epilepsy, before permanent changes can occur in neuronal aggregates that promote hyperexcitability and seizure spread. The causative role of inflammation in the pathogenesis of epilepsy and neurological sequelae of chronic intractable epilepsy needs to be proven and requires further investigation from both clinical and basic science angles.

 XML Download
XML Download