

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The production of new forms of manufactured/engineered nanoparticles is of increasing concern as nanotechnology continues to develop and manufacture them.1 These form a plethora of particle types that include nanotubes, fullerenes, quantum dots and compound particles of various types. Whilst information on the toxicity of new types of nanoparticles (NP) is accumulating these are mostly in vitro studies, with few animal or human studies. The existing toxicology knowledge regarding NP is almost entirely based on combustion-derived nanoparticles (CDNP) present in environmental air. Evolving from the 'ultrafine hypothesis',2,3 this strand of research has focused on CDNP like diesel soot since this component of particulate matter (PM) is seen as a key component mediating adverse health effects. The mechanism at the cellular level is understood in terms of the ability of particles to cause oxidative stress and inflammation and translocate from the site of deposition.4 This review builds on environmental NP and their mechanisms, as a basic paradigm and then moves on to discuss toxicology of engineered NP.
The portal of entry for NP discussed here is the lungs and the toxic effects seen there are discussed. In addition the lungs and the cardiovascular system are intimately linked and the PM10 literature indicates clearly that the cardiovascular system is a lead target system for the adverse effects of PM.
COMBUSTION-DERIVED NANOPARTICLES IN ENVIRONMENTAL AIR POLLUTION
PM10 and its adverse effects
The adverse health effects of air pollution have been recognised throughout much of recorded time and are now documented in large international epidemiological studies. In the UK, fossil fuel combustion in towns and cities, during periods of cold weather, where there is little mixing of air have been associated with the generation of smog episodes. These smogs consisted largely of sulphur dioxide and particles and could very high concentrations in urban air. The particle component or PM represents a key part of the air pollution cocktail present in ambient air, which also comprises gases such as ozone, nitrogen dioxide etc. PM in ambient air is measured as the mass of particles collected using the PM10 or PM2.5 sampling conventions.5 The adverse health effects of PM are seen at the levels that pertain in UK and other cities today and there is often no threshold. In other words there is a background of ill health being caused by PM that increases when the ambient particle cloud increases in concentration and goes down when the amount of particles in the air decreases.6
These adverse health effects of air pollution have been measured in hundreds of studies and there is good coherence between the acute effects seen in time series and panel studies, and the chronic effects seen in environmental studies.
Nanoparticles as the most toxic component of PM10
PM is a complex mixture of particle types that depend on season, time of day, site of sampler etc. CDNP are present in PM from conurbations and are a major toxicologically important component. CDNP originates principally from car exhausts although there are other sources.4 Sulphates tend to be very low in toxicity in experimental studies, 7 but do show a relationship with adverse effects in some epidemiological studies;8 this apparent anomaly may be explained by a correlation between sulphates and some more potent component of the air pollution mix which is actually driving the adverse effect, such as fine particles.
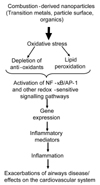
NP number, likely to be principally comprised of CDNP, ranged from 15,000 to 18,000 particles per cm3 in 3 European cities9 and 10,000 to 50,000 particles per 3 in a busy London street.10 In a study on US highways exposure in a vehicle travelling in busy traffic was reported to be 200 to 560 × 103 particles per cm3, (predominantly NP).11,12 Indoor air also contains NP and cooking, vacuuming and burning wax candles produce NP of soot.13 NP also produced during combustion of domestic gas and in one study 3 gas rings produced around 50,000 particles per cm3 which underwent rapid aggregation within a few minutes, as evidenced by increases in particle size and decrease in apparent number.14 Secondary NP also arise from environmental chemistry, e.g. nitrates; but these are unlikely to be as toxicologically potent as CDNP (see below). The molecular mechanisms of the adverse effects of CDNP has been extensively reviewed by the authors4,15,16 and the pro-inflammatory mechanism is summarised in Fig. 1.
CDNP AND THE LUNGS
The present understanding of CDNP activity in the lungs is that the surfaces, organics and metals can all produce free radicals with the potential to produce oxidative stress and contribute to inflammation. Diesel exhaust particles (DEP) are one of the main CDNP to which individuals are exposed. DEP causes inflammation in rat17,18 and human lungs19 following short-term, high level exposure. Oxidative stress is demonstrable as increased levels of 8-OHdG, the oxidative DNA adduct of the hydroxyl radical, in the lungs of rats following exposure and in cells in culture treated with DEP.20,21 The component of DEP responsible for the oxidative stress and subsequent pro-inflammatory signalling is principally the organic fraction,22-25 although transition metals may also be involved especially for welding fume.26 The oxidative stress then causes activation of signalling pathways for pro-inflammatory gene expression, including MAPK22,27-29 and NF-κB activation22,30 and histone acetylation that favours pro-inflammatory gene expression.31 Activation of these pathways culminates in transcription of a number of pro-inflammatory genes such as interleukin-8 (IL-8) in epithelial cells treated in vitro32 and in human lungs exposed by inhalation.33 Tumour necrosis factor-alpha (TNF-α) has been reported to be increased in macrophages exposed to DEP in vitro34 and interleukin-6 (IL-6) is released by primed human bronchial epithelial cells exposed to DEP.35
CARDIOVASCULAR EFFECTS OF PM AND CDNP-POTENTIAL EFFECTS ON ENDOTHELIUM AND ATHEROSCLEROTIC PLAQUE STABILITY
The adverse cardiovascular events associated with increases in PM36,37 could be mediated through the effects of CDNP. In a mixture of studies which have used PM, CAPS and model NP, evidence is accumulating that NP cause inflammation that could adversely affect the cardiovascular system. There is evidence of systemic inflammation following increases in PM, as shown by elevated C-reactive protein, blood leukocytes, platelets, fibrinogen and increased plasma viscosity (reviewed in reference 38). Atherothrombosis is the principle cause of cardiovascular morbidity and mortality.39 Atherosclerosis is an inflammatory process, initiated via endothelial injury and producing systemic markers of inflammation that are risk factors for myocardial and cerebral infarction.39-41 Repeated exposure to PM10 may, by increasing systemic inflammation, exacerbate the vascular inflammation of atherosclerosis and promote plaque development or rupture. Experimental studies with animal models susceptible to atherosclerosis confirm the ability of particle exposure to enhance atherosclerosis.42,43
Normally the endothelial monolayer delicately balances regulatory pathways controlling vasomotion, thrombosis, cellular proliferation, inflammation and oxidative stress. Endothelial dysfunction or denudation is one of the earliest pathological features of atherosclerosis.44 Loss of endothelial function results in expression of leukocyte adhesion proteins, reduced anticoagulant activity and the release of growth factors, inflammatory mediators and cytokines. Chronic inflammation results in leukocyte and monocyte recruitment, induction of atheroma formation and further arterial damage. Plaque expansion and disruption can lead to angina, crescendo angina and acute coronary syndromes, including myocardial infarction.44-46
Inhaled PM may influence the vasculature through indirect effects mediated by pulmonary inflammation or through the direct action of particles that have become blood-borne. Whether inhaled NP can access the circulation is currently the subject of intense research47-49 and there is conflicting reports on whether Technegas-radioactive carbon NP-can reach the blood following inhalation in humans.50,51 Certainly, injured arteries can take up blood borne NP, a fact exploited by the nanotechnology industry for both diagnostic and therapeutic purposes in cardiovascular medicine. The intra-arterial infusion of carbon black NP has a detrimental effect on the mouse microcirculation with up regulation of von Willebrand factor expression and enhanced fibrin deposition on the endothelial surface.52 These prothrombotic effects are in keeping with toxicological evidence from instillation studies, which suggest particle exposure may promote thrombogenesis.53,54
The endothelium plays a vital role in the control of blood flow, coagulation, fibrinolysis and inflammation. Following the seminal work of Furchgott and Zawadski,55 it is widely recognised that an array of mediators including cigarette smoking can influence vascular tone through endothelium-dependent actions, and there is now extensive evidence of abnormal endothelium-dependent vasomotion in patients with atherosclerosis.56-58 Mild systemic inflammation also causes a profound, but temporary suppression of endothelium-dependent vasodilatation.59 However, whilst endothelium-dependent vasomotion is important, it may not be representative of other aspects of endothelial function, such as the regulation of fibrinolysis.
The fibrinolytic factor tissue plasminogen activator (t-PA) regulates the degradation of intravascular fibrin and is released from the endothelium through the translocation of a dynamic intracellular storage pool.59,60 If endogenous fibrinolysis is to be effective, then the rapid mobilization of t-PA from the endothelium is essential because thrombus dissolution is much more effective if t-PA is incorporated during, rather than after, thrombus formation.61,62 The efficacy of plasminogen activation and fibrin degradation is further determined by the relative balance between the acute local release of t-PA and its subsequent inhibition through formation of complexes with plasminogen activator inhibitor type 1 (PAI-1). This dynamic aspect of endothelial function and fibrinolytic balance may be directly relevant to the pathogenesis of atherothrombosis.
CDNP and endothelial dysfunction
In order to investigate the potential role of the endothelium in triggering of acute myocardial infarction following CDNP exposure we investigated the effects of diesel exhaust inhalation on vascular and endothelial function in humans.63 In a double-blind, randomized, cross-over study, 30 healthy men were exposed to diluted diesel exhaust at 300µg/m3 particulate, or air, for 1 hour with intermittent exercise. Two and six hours after exposure, bilateral forearm blood flow was measured following infusions of the endothelium- dependent vasodilators bradykinin (BK) and acetylcholine (ACh), and endothelium-independent vasodilators sodium nitroprusside (SNP) and verapamil. Inflammatory mediators in blood were measured concomitantly. We showed no differences in resting forearm blood flow or inflammatory markers after exposure to diesel exhaust or air. There was a dose-dependent increase in blood flow with each vasodilator, but this vasomotor response was significantly attenuated to BK, ACh and SNP (p < 0.001) infusions 2 hours after exposure to diesel exhaust, and remained impaired at 6 hours.
In addition, BK caused a dose-dependent increase in plasma t-PA that was suppressed 6 hours after exposure to diesel (p < 0.001; area under the curve decreased by 34%). We concluded that, at levels encountered in an urban environment, inhalation of dilute diesel exhaust impairs two important and complementary aspects of vascular function in humans: the regulation of vascular tone and endogenous fibrinolysis. The likely mechanism- is shown in Fig. 2 and is based on the central role of nitric oxide (NO) in the maintenance of vascular tone.
The vasodilator drugs ACh and BK act on receptors in the endothelium to stimulate calcium increase and endothelial nitric oxide synthase (eNOS) activation leading to local nitric oxide (NO) levels that activate gunayl cyclase in subjacent smooth muscle cells stimulating relaxation. SNP is a NO donor and increases vascular NO directly by an endothelial-independent pathway, while Verapamil causes smooth muscle relaxation by an endothelium-independent and NO-independent pathway. The observed blunting of the vasomotor response to ACh, BK and SNP could be explained by oxidative stress from pulmonary inflammation or from particles that gain access to the blood. In this scenario, superoxide anions in the vessel wall, resulting from oxidative stress, rapidly combine with NO to form peroxynitrite. Therefore less NO is available in the smooth muscle cells and relaxation is blunted.
This explanation combines the well-known oxidative stressing and inflammatory effects of CDNP with key endothelial functions and so provides a potential mechanism that links air pollution to the pathogenesis of atherothrombosis and acute myocardial infarction.63
Engineered nanoparticles
One of the major reasons behind the rapid expansion in industrial use of nanotechnology and in particular engineered NP themselves, are their unique properties due to small size and large reactive surface area. NP come in a wide variety of shapes, sizes and chemical compositions. In addition to the spherical shapes observed for particles such as titanium dioxide (TiO2), shape varieties also include carbon nanotubes, nanowhiskers and nanofibres. Engineered NP vary considerably in their size and composition and so would be anticipated to vary in toxicity. Nanotubes and nanowires can range from less than 100 nm diameter to tens of µm in length. The variety of chemical composition range from substances considered traditionally to be relatively inert (e.g. carbon and gold) to substances associated with significant toxicity (e.g. cadmium and other heavy metals). Since the NP size imparts heightened reactivity to the 'inert' materials, it is interesting to consider the impact of small size on toxic materials, especially since reactivity might relate to toxicity.
Engineered nanoparticles and the lungs
Carbon black (CB) and TiO2 along with alumina and silica have been studied for some time with regard to their pro-inflammatory effects but none of these studies have explicitly addressed effects on the cardiovascular system. Nano-size CB has been intensively studied with regard to the issue of low toxicity dust and the confounding effect of rat lung overload.64-67 In view of the very high surface area per unit volume of NP and the identification of surface area as the driver for overload,68-70 attention has been focused on the nanoparticulate form of these nuisance dusts. These would be anticipated to produce lung overload at lower mass lung burdens than seen with the larger particles and in fact, was indeed shown to be the case.70,71 However, even at low, non-overload exposures to nano-sized CB, there was a pro-inflammatory effect not seen with the larger CB particles.72 Instillation studies have also shown that the nanoparticulate form of CB and TiO2 produce more inflammation than an equal mass of larger, yet respirable particles73,74 of the same material and across a range of NP of nuisance dust the surface area was found to be the driver of the inflammation.75
The molecular mechanism of the increased inflammatory effects of nanoparticle CB have demonstrated that they generate reactive oxygen species (ROS) in cell-free systems76-78 and cause alterations in calcium signaling79,80 in exposed cells. Oxidative stress from the nanoparticulate CB can also activate the EGF-receptor81 and redox-responsive transcription factors such as NF-κB80 and AP-182 leading to the transcription of pro-inflammatory cytokines and lipid mediators.78,80
Carbon nanotubes
Carbon nanotubes (CNT) are long sheets of graphite rolled in the form of a tube, that can range from a few nm thick (single-walled-SWCNT) up to a few hundred nm thick (multi-walled-MWCNT). The needle-like structure implies that a paradigm related to fibres, such as asbestos, might be appropriate in considering their toxicity. The potential pathogenicity of a conventional fibre is dictated by length greater than 20µm, thinness and biopersistence.83 Biopersistence is an important determinant of mineral fibres and Synthetic Vitreous Fibre pathogenicity. Long biopersistent fibres are the biologically effective dose that drives pathogenic effects83 whilst non-biopersistent fibres undergo dissolution processes that can be enhanced at the acid pH of 5.0 existing inside macrophage phagolysosomes.84 Long, biosoluble fibres undergo leaching of key structural molecules leading to breakage into short fibres that are readily phagocytosed by macrophages.83,85
In one study,86 the authors addressed the important issue of biopersistence of CNT using both un-ground and ground nanotubes, 0.7 and 5.9µm long respectively. These were assessed for biopersistence and the longer, un-ground nanotubes were more biopersistent than the short ones. This is consistent with the greater biopersistence of long fibres seen in studies with asbestos and other mineral fibres although these 'long' nanotubes were much shorter than those mineral fibres defined as 'long', which are in the region of 20µm and greater.83 A 20µm diameter rat macrophage is able to enclose and transport fibres less than its own diameter from the lungs,83 and the length-dependent inhibition of clearance seen with 5.9µm long nanotubes is thus rather unexpected. It may be that the well-documented tendency for nanotubes to form bundles and wires87 is important in impairment of clearance.
Nanotubes have been used in a number of rat lung instillation studies.83,88-90 All of these used high dose and dose-rate which raises questions about physiological relevance; no study has addressed the role of length by comparing long (> -20µm) with short (< 10µm) CNT. However all of the studies mentioned above showed an increased ability of CNT to cause granulomatous fibrosis in the absence of severe inflammation.
CNT have also been tested in a range of different cell types in vitro to assess their potential toxicity. Treatment of human keratinocytes have shown that both SWCNT and MWCNT are capable of being internalized, causing cellular toxicity.91,92 In a study with alveolar macrophages, SWCNT were more cytotoxic than MWCNT after exposure at equal mass dose.93 Human T cells exposed to oxidised MWCNT were killed in a time- and dose-dependent manner, through mechanisms involving apoptosis94 as was also the case in kidney cells exposed to SWCNTs.95 Manna et al., demonstrated dose-dependent oxidative stress and NF-κB activation in human keratinocytes along with IκB depletion and MAPK phosphorylation.96 In vitro, CNT can produce free radicals by the role of iron via Fenton-type reactions.97
It is difficult to draw general conclusions on CNT toxicity because of the scarcity of data and CNT variability-they can vary in length and composition including metal contamination. CNT are often kinked and tangled into aggregates of varying size and shape. This kind of variability is found both between and within samples. All of these factors could impact on toxicity. More rigid CNT are likely to disperse more efficiently than tangled CNT. However the tangles are more easily taken up by cells in culture and could therefore be more readily cleared from the lungs by macrophages. A programme of research is warranted to define the factors that control CNT toxicity.
Engineered NP and the cardiovascular system
Radomski et al., examined the role of new NP on the clotting system98 studying the effects of multi-walled and single-walled nanotubes, C60 fullerenes and mixed carbon black NP on human platelet aggregation in vitro and rat vascular thrombosis in vivo. Standard urban PM was used as a control. Nanotubes and carbon black particles but not C60, stimulated platelet aggregation and the same ranking was observed in their ability to affect the rate of vascular thrombosis in rat carotid arteries; urban dust had low activity in these assays. Thus, there are differences between different carbon NP to activate platelets and enhance vascular thrombosis. Yamawaki et al., have shown that carbon back of aggregate size 248nm are cytostatic, cytotoxic and pro-inflammatory in endothelial cells.99
Engineered NP and Quantitative Structure Activity Relationships
The numbers of new NP that are being produced pose a special problem in testing. NP are relatively easy to alter in terms of physicochemistry, size, coating, composition etc and this makes for even more particles to be tested. Other factors are also important-the average small company that is developing a new NP type is unlikely to have the funds to carry out proper toxicology testing and the current climate against animal testing makes this type of testing not viable.
In the pharmacology and toxicology worlds, the term QSAR (Quantitative structure activity relationship) is used to describe the attempt to relate chemical structure to pharmacological or toxicological activity. This idea could be used to categorise NP on the basis of physicochemistry, if physicochemical markers could be related to toxicity. The most obvious candidates for structural markers that could be related to toxicity markers at the moment are size/surface area and oxidative stress. For insoluble particles the surface area times the surface reactivity describes a biologically effective dose (the dose that drives adverse effects) and so particle size is likely to be important. For NP, the quantum effect changes in the physicochemical nature of the surface of a very small particle compared to a larger one and could plausibly impact on toxicology. The dominant hypothesis for the action of harmful particles on cells is oxidative stress and the oxidative stressing activity of particles is a physicochemical parameter that may well be important in structure activity considerations.
The QSAR idea is likely to be achievable for predicting lung inflammation and so could be predictive for cardiovascular effects if they are driven by pulmonary inflammation. For translocation from the lungs to the blood or for effects in the blood, a different QSAR may be necessary.
Potential carcinogenic effects of manufactured NP
The ability of PM to cause cancer is well documented and other types of particles, such as asbestos and silica, are also known to be carcinogenic. The mechanisms are, however, not completely understood and may involve both direct genotoxic effects of the particles themselves and indirect genotoxic effects mediated through the particles ability to cause inflammation. Direct genotoxic effects of particles involve the particles entering cells and delivering damage to DNA. The chemical composition, as well as the structural composition of the particles, both plays a role in this and so CDNP certainly have the potential to mediate this type of effect.100 Transition metals have been shown to redox cycle inside the cell and generate damaging hydroxyl radicals that form mutagenic adducts with DNA.101 Organic molecules adsorbed on to the surface of CDNP, such as polycyclic aromatic hydrocarbons (PAHs), can also form adducts102 whilst large surface areas on NP are capable of generating oxidative stress. 100 In addition, the inflammatory effects of CDNP, as discussed above, can play an important role in the genotoxic and carcinogenic processes and the products of the leukocyte oxidative burst can form adducts within target cells.103 The effects of oxidative stress inside the cell may cause lipid peroxide production in the cell and the products of lipid peroxidation are longer-lived than the ROS themselves and may therefore mediate adduct formation.104
Certain types of NP appear to be able to enter the nucleus in cell culture systems, to a much greater extent than larger particles of the same material105 and NP in general seem to be cable of crossing biological membranes.106 If NP generally gain access to the nucleus rather than being retained in the cytoplasm like larger particles, then by virtue of being closer to the DNA, the oxidative products they produce may be more likely to cause genotoxic effects.
CONCLUSION
A paradigm has evolved arising from experience with environmental CDNP, exemplified by diesel soot. In this paradigm, oxidative stress and inflammation are identified as key processes in the local effects in the lungs. In addition, inflammatory effects and blood translocation could explain adverse cardiovascular effects observed in epidemiology studies with air pollution particles. Support for this contention comes from a number of studies using model NP and CDNP, where adverse cardiovascular effects such as clotting, plaque development and endothelial dysfunction are enhanced after NP exposures in a number of different models. In parallel with these studies, an increasing number of toxicology studies using bulk NP, such as TiO2 and CB, have identified a key role for the large surface area of NP and its ability to produce oxidative stress.78,107,108 It is not known whether the same paradigm can be used for new engineered NP and nanotubes. In the limited studies so far published, engineered NP, such as the CNT are also reported to induce oxidative stress, cell death and inflammation. However, there are differences in the magnitude of the adverse effects caused by NP between models and not all NP are likely to have the same toxic potency. This is to be anticipated since the total toxicity of any particle sample is the complex sum of the surface reactivity times the surface area plus releasable toxic moieties, along with shape, and all modified by the degree of biopersistence. There is a strong likelihood that these factors will differ considerably and so the cumulative toxicity will vary between different particle types.
 XML Download
XML Download