

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hypoglycemia is a heterogenic metabolic syndrome with a complicated pathogenesis. Ketotic hypoglycemia is the most common type of hypoglycemia in childhood. However, hyperinsulinism, hypopituitarism, or hereditary hepatic enzyme deficiencies are considered in patients with persistent or refractory hypoglycemia (1).
Insulinoma is the most common cause of hyperinsulinemic hypoglycemia (1-4/million patients) and the second most common (10%-30%) functioning pancreatic islet cell tumor associated with multiple endocrine neoplasia type 1 (MEN1) (2,3) after gastrinoma. In contrast, only 4%-6% of patients with insulinoma will develop MEN1 (4-6). Unlike sporadic insulinomas that usually develop after the age of 40, MEN-associated insulinomas usually occur before 40 years of age and even sometimes before 20 (7,8). Nevertheless, there are rare cases of insulinoma presenting as the first sign of MEN1 in the pediatric population.
MEN is characterized by the occurrence of a tumor involving two or more endocrine glands within a single patient and has an equal sex distribution (9,10). MEN type 1 (MEN1) occurs in approximately one in 30,000 individuals. It encompasses tumors of the parathyroids (95% of cases), pancreatic islets (30%-80%), and anterior pituitary (15%-90%). We report a girl with a newly diagnosed MEN1 who presented with hypoglycemia as initial presentation.
CASE DESCRIPTION
In May 2013, a 9-year-old female presented with an episode of sudden loss of balance at home and some tremor. She had experienced occasional seizure like movements early in the morning for the past 3 months. She also had dizziness and difficulty waking up in the morning for 6 months. The symptoms disappeared after eating. The patient therefore ate more than usual and gained 10 kg within the previous year. She was not taking any prescription medications at the time of her evaluation. On the day of the hospital visit, she had experienced generalized tonic-clonic seizure that lasted for 5 minutes early in the morning.
Physical examination revealed a well-nourished girl with normal vital signs. She weighed 41 kg with body mass index (BMI) of 22.5 kg/m2. She complained of dizziness, occasional palpitation, weakness, hunger, and sweating. Her abdomen was soft and non-tender without palpable masses or organomegaly. Neurologic examinations were normal.
The serum glucose level measured at the emergency department was 34 mg/dL. Laboratory evaluation showed elevated insulin of 142.7 uIU/mL and elevated C-peptide at 5.88 ng/mL. Her serum PTH level was 15 pg/mL (normal range, 11–62 pg/mL), serum IGF-1 was 298 (normal range, 78–517 ng/mL), serum prolactin level was 7.5 ng/mL (normal range, 2.7-19.7 ng/mL) and serum calcium level was 9.3 mg/dL (normal range, 8.8-10.8 mg/dL). She received intravenous glucose and her symptoms were promptly resolved. We then began to check for diseases that could cause hyperinsulinemic hypoglycemia and conducted an image work-up to rule out insulinoma.
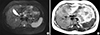
Magnetic resonance study of the pancreas revealed a 1.3 × 1.5 cm nodule with high signal intensity on T2 weighted images, which appeared as a well circumscribed mass at the head of the pancreas and with hyposignal intensity on T1 weighted images (Fig. 1). We suspected insulinoma and performed hepatic venous sampling, collecting 8 samples at the splenic vein, the superior mesenteric vein, and the portal vein. The insulin levels for the 8 samples were 127.1, 126.7, 115.2, 118.4, 251.6, 129.8, 458.6, and 199.4 uIU/mL, with the highest levels in the portal vein.
Fig. 1
MR study of the pancreas. (A) A mass with hyposignal intensity on T1 weighted images, 1.3 × 1.5 cm. (B) High signal intensity on T2 weighted images at the head of the pancreas.
The patient underwent pancreatic exploration with enucleation of the pancreatic mass. Histopathological evaluation of the pancreatic mass was consistent with insulinoma, specifically, a well differentiated pancreatic insulinoma with a grandular growth pattern and a fibrous stroma.
Immediately after removal of the mass, the patient’s glucose level increased to 152 mg/dL. Post-operative glucose levels were consistently over 100 mg/dL and she experienced no further hypoglycemic episodes. The patient was discharged in good health with proper glucose level.
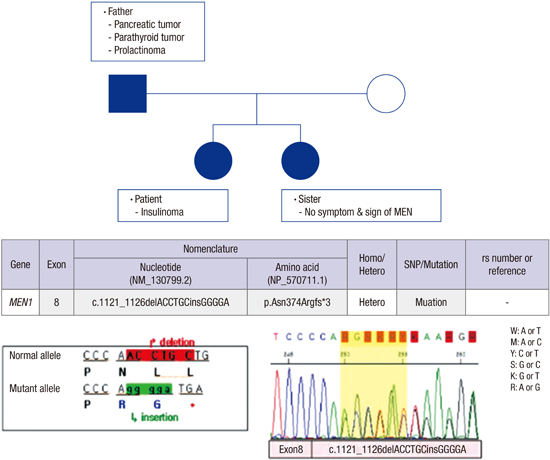
Four months after the patient’s hospital visit, her father had a health checkup due to occasional hypoglycemia and was diagnosed with pancreatic tumor, parathyroid tumor and prolactinoma. Based on his daughter’s history and the presence of three different tumors, he was suspected of MEN. DNA sequencing analyses revealed that not only did the father have a mutation in the MEN1 gene, the patient and patient's younger sister also had the same mutation (Fig. 2). However, the younger sister had no symptoms or signs of MEN1. We have monitored the patient with regular follow-ups at the out-patient department for a year and have not detected any abnormal signs or symptoms.
Fig. 2
Pedigree and DNA sequencing analysis. MEN 1 mutation. (c.1121_1126delACCTGCinsGGGGA) was found in the patient, patient's father and younger sister.

Ethics statement
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional review board. The institutional review board of the Ajou University Hospital reviewed and approved the present genetic study (AJIRB-MED-EXP-15-109). The board waived informed consent.
DISCUSSION
There are two different forms of MEN1: familial and sporadic (9). Most cases of MEN1 are the familial form, which is inherited as an autosomal-dominant disorder. The sporadic form develops in 8%–14% of patients with MEN1 and molecular genetic studies have confirmed the occurrence of de novo mutations of the MEN1 gene about 10% of patients with MEN1 (11,12).
A diagnosis of MEN1 may be established in an individual that fulfills one of three criteria. MEN1 may be clinically diagnosed in an individual based on the occurrence of two or more MEN1-associated endocrine tumors (4,13). A diagnosis of familial MEN1 can be established in individuals with one of the MEN1-associated tumors who are first degree relatives of patients with a clinical diagnosis of MEN1. Finally, a genetic diagnosis of MEN1 is made upon identification of a germline MEN1 mutation in an individual who may be asymptomatic and has not yet developed any of the serum biochemical or radiological abnormalities indicative of tumor development (13). In our case, genetic study of MEN1 was not performed because insulinoma was the only manifestation and there was no familial history at the first evaluation. However, her father’s diagnosis led to additional DNA sequencing analysis, resulting in the diagnosis of MEN1 in the patient.
An electronic search of the PUBMED database was conducted using the keywords insulinoma and MEN1. The search was limited to children and all abstracts were screened for insulinoma with MEN1, revealing only four pediatric cases. The minimum age of onset was 8 for MEN1 insulinoma (age range 8-14 years) (14). Most of the insulinomas were located within the pancreas and were treated with surgery. Our case was consistent with previous cases of pediatric MEN1, in which patients diagnosed with hyperinsulinism-related hypoglycemia presented with loss of consciousness, seizure or syncope (14-17) and were treated with surgery.
In the previously reported case, one patient died due to multiple metastases. Metastases occur in up to 50% of patients with MEN1-associated insulinomas, compared to 10% of non-MEN1 insulinomas, in all age groups (3,18).
The international guideline of management and therapy in MEN1 mutations recommends a biochemical evaluation of prolactin serum concentration from age 5, a biochemical screen of fasting total serum calcium concentration (corrected for albumin) from age 8, a biochemical screen of fasting serum gastrin concentration from age 20, magnetic resonance imaging (MRI) of the head every 3–5 years from age 5 and abdominal CT or MRI every 3–5 years from age 20 in children diagnosed with MEN1 mutation (9). A biochemical screening for the fasting serum concentration of full-length PTH, yearly chest CT, yearly somatostatin receptor scintigraphy and yearly octreotide scans are also recommended.
In conclusion, this case reports MEN1 mutation in a 9-year-old girl that showed insulinoma as the first manifestation. Early clinical and genetic identification of affected individuals is helpful for monitoring them and also for genetic counseling even if the patient shows only one manifestation that is not insulinoma.
 XML Download
XML Download