

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Gaucher disease (OMIM #230800) is an autosomal recessive disease caused by a β-glucocerebrosidase deficiency, and characterized by hepatosplenomegaly, anemia, thrombocytopenia, bone pain, and growth retardation (1). β-glucocerebrosidase degrades glucocerebroside into glucose and ceramide, and loss of function leads to accumulation of glucocerebrosides in various tissues including the liver, spleen, central nervous system, skeletal system, and lungs (2). Gaucher disease can be categorized into three subtypes according to the presence of neurological deterioration, age at diagnosis, and rate of progression, namely, non-neuronopathic (type 1), acute neuronopathic (type 2), and chronic neuronopathic types (type 3) (3). Type 1 Gaucher disease is the most prevalent form and is characterized by an absence of neurologic symptoms.
Enzyme replacement therapy (ERT) remarkably improves the clinical outcome of patients with Gaucher disease, particularly hepatosplenomegaly and hematological abnormalities (4). Aglucerase (Ceredase®, Genzyme Corp., Cambridge, MA, USA), a mannose-terminated placental-derived β-glucocerebrosidase, was approved by the Food and Drug Administration (FDA) for treatment of type 1 Gaucher disease in 1991 (5). Similarly, imiglucerase (Cerezyme®, Genzyme Corp.), a recombinant human β-glucocerebrosidase, was approved by the FDA in 1994. Imiglucerase can be produced in large quantities in Chinese hamster ovary (CHO) cells via recombinant technology, and used the macrophage mannose receptor to deliver the enzyme to lysosomes (6). Imiglucerase is considered the prototype for ERT in lysosomal storage diseases, and the long-term safety and efficacy of imiglucerase has been well validated (7). Currently, two other forms of recombinant β-glucocerebrosidase have been developed for treatment of Gaucher disease: velaglucerase alfa (Shire Human Genetic Therapies, Lexington, MA, USA) and taliglucerase alfa (Pfizer, New York, NY, USA) (8, 9). Despite having different oligosaccharide modifications, their therapeutic efficacy is similar (7, 10).
Unfortunately, the high cost and global shortage of imiglucerase has been problematic for patients and health care providers (11, 12). However, a biosimilar of imiglucerase, Abcertin® (ISU302, ISU Abxis, Seoul, Korea), has recently been developed. Therefore, this study was performed to evaluate the short-term efficacy and safety of Abcertin® in patients with type 1 Gaucher disease who were previously treated with imiglucerase.
MATERIALS AND METHODS
Subjects
A total of six patients with non-neuronopathic Gaucher disease were screened for enrollment. All patients were diagnosed with Gaucher disease by a documented deficiency of leukocyte glucocerebrosidase activity, and mutation analysis of the GBA gene using genomic DNA from peripheral blood leukocytes. Polymerase chain reactions of all coding exons and exon-intron boundaries were performed with allele-specific primers, and direct sequencing of amplification products was performed in both the forward and reverse directions using an ABI3130x1 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
The inclusion criteria for this study were as follows: 1) patients diagnosed with type 1 Gaucher disease; 2) patients who were stable with Cerezyme® therapy and who were maintained on a dose of Cerezyme® for at least 6 months prior to enrollment; 3) patients older than 2 yr of age; and 4) nonpregnant female patients. All these patients were stable following treatment with imiglucerase. Stable treatment was defined as an absence of neurologic deficits, normal hemoglobin levels, platelet counts greater than 100,000/µL, normal or an absence of deteriorated bone mineral density, and an absence of aggravated splenomegaly or hepatomegaly.
We excluded patients who exhibited fluctuating hemoglobin or platelet levels (at least six months prior to the study), hypersensitivity to imiglucerase (Cerezyme®), anemia due to iron, folic acid, or vitamin B12 deficiency and clinical findings of type 2 or 3 Gaucher disease. Patients who received miglustat, erythrocyte growth factor, or systemic corticosteroids during the six months prior to enrollment were also excluded.
Study drug; Abcertin®
Abcertin® (ISU302, ISU Abxis, Seoul, Korea) is a recombinant protein produced by genetically engineered CHO cells. Abcertin® consists of 497 amino acids with two disulfide bridges and four N-glycosylation sites, together with a terminal mannose exposed by multi-step digestion. Abcertin® has a similar amino acid sequence to human glucocerebrosidase with only a single amino acid difference (arginine replaced with histidine at the 495th amino acid). The imiglucerase was produced in serum-free medium using suspension cell culture technology and was purified a series of chromatography steps. The structural, physicochemical, immunological and biological properties of imiglucerase have been well characterized both in vivo and in vitro.
Study design
The study was an open-label, switch-over study from Cerezyme® to Abcertin® (ISU302). Abcertin® was administered to each subject at a dose of 30-60 U/kg every two weeks. Abcertin® (30-60 U/kg) was diluted with 0.9% saline solution for injection at a final volume of 100-200 mL. At the first visit, the study drug was administered intravenously at a rate of ≤1 U/kg/min for approximately 90±3 min for pharmacokinetic assessments. At the second or later visits, the study drug was administered for 60 min or longer. For three subjects, the study period was extended for another 6 months because of a supply shortage of Cerezyme®. The 6-month extension was approved by the Korea FDA.
Pharmacokinetic assessment
Pharmacokinetic assessments were performed during the first infusion. Blood samples were collected at nine time points (before infusion, and at 15, 30, 60, 90, 100, 120, 150, and 180 min), and β-glucocerebrosidase activity was measured. Blood concentration time courses are shown as a linear or log/linear graph for each subject. Cmax and Tmax were measured and the area under the curve (AUC) was calculated using a linear up/log down method. Parameters were calculated using Phoenix WinNonlin Version 6.2 (Pharsight, CA, USA) software and standard noncompartmental methods.
Subject group for efficacy and safety analysis
Subjects were classified into three groups, as follows: 1) a modified intention-to-treat (MITT) group consisting of subjects who underwent primary efficacy assessment one or more times after drug administration; 2) a per-protocol (PP) group consisting of subjects included in the MITT who did not violate the inclusion/exclusion criteria and who were administered the study drug for at least 60% of the total 48 weeks. Subjects who completed less than 60% of the study or withdrew were excluded from the PP group; and 3) an intention-to-treat (ITT) group consisting of all subjects who received the study drug.
Assessment of efficacy
Primary efficacy endpoints were hemoglobin concentration and platelet counts before and during the study period. Secondary endpoints for efficacy assessment included reduced spleen and liver volumes determined from volumetric computerized tomography (CT), changes in liver function (serum aspartate transaminase [AST] and alanine transaminase [ALT] levels), skeletal abnormalities, bone mineral density (BMD), and serum biomarkers (acid phosphatase, angiotensin converting enzyme [ACE], and chitotriosidase levels) before and during the study period (13).
Hemoglobin concentrations, platelet counts, serum AST, and ALT levels were measured every 6 weeks. Serum acid phosphatase, ACE, and chitotriosidase levels were measured every 12 weeks during the study period. Liver and spleen volumes were measured by CT at 0, 24, and 48 weeks (where applicable). Liver and spleen volumes were measured in mL and calculated as a percentage of body weight to allow comparison between patients of different ages; values were also expressed as multiples of normal (MN) at baseline and week 24. A normal spleen volume was considered 0.2% of body weight, and a normal liver volume 2.5% of body weight in both children and adults, respectively (14). Skeletal status was evaluated by simple X-ray and assessed as none, mild, moderate, or severe for osteosclerosis and osteonecrosis, respectively, which was then expressed as 0, 1, 2, and 3 points (15). BMD of the femur neck and lumbar spine (L2-4) were measured by dual energy radiography absorptiometry (Lunar Corp., Madison, WI, USA) at 0, 24, and 48 weeks (where applicable). Age- and sex-matched BMD Z-scores for Korean children and adolescents were used for comparisons of BMD during treatment (16).
Assessment of safety
Vital signs were measured initially and every 2 weeks after administration of the study drug, and changes were analyzed. Treatment-emergent adverse events (TEAE) were recorded from initiation of study drug administration to 2 weeks after the last administration. In addition, all adverse events that occurred after drug administration were classified according to their severity level and relevance to the study drug, and summarized as System Organ Class and Preferred Term using the Medical Dictionary for Regulatory Activities. An adverse drug reaction was defined as a responsible event associated with drug infusion, and the relationship to drug infusion was judged by the investigator.
Immunogenicity tests were performed every 12 weeks in all patients and consisted of screening for IgG antibodies to imiglucerase. Anti-imiglucerase antibodies in plasma were detected by an enzyme-linked immunosorbent assay.
Statistical analysis
All statistical analyses were performed using SPSS (SPSS for Windows, version 21.0, SPSS, Chicago, IL, USA). For efficacy assessments, mean differences between baseline and treatment levels were analyzed using the Wilcoxon signed rank test. Differences in biochemical parameters over time were also analyzed for significance using a repeated measured ANOVA. The McNemar test was used to test changes in antibody formation before and after drug administration. P values below 0.05 were considered statistically significant.
Ethics statement
This study was approved by the institutional review board of the Asan Medical Center, Seoul, Korea (IRB No. 2013-0848) and written informed consent was obtained from all subjects or parents. This study was registered at www.clinicaltrials.gov as NCT-02053896.
RESULTS
Baseline characteristics of subjects
Out of the six patients who underwent screening, one patient was withdrawn due to ineligibility, and the remaining five (three males and two females) were enrolled. The mean age of the subjects was 16.2±8.26 yr (range, 8-29 yr). The mean disease duration from the date of diagnosis to the date of enrollment was 104±48.16 months (range, 68-183 months). The subjects previously received Cerezyme® at a dose of 30-55 units/kg once every 2 weeks (Table 1).
For the five subjects, no serious violations of the study protocol that could potentially affect the safety and efficacy of Abcertin® were found during the study period, and no subjects were withdrawn due to adverse events, absence of efficacy, or voluntary withdrawal from participation. All five patients were included in the ITT group for safety assessment, and the MITT and PP groups for efficacy assessment.
Pharmacokinetics of Abcertin®
Pharmacokinetic profiles were assessed after Abcertin® was intravenously administered for 90 min. Abcertin® had a Tmax of 1.34±0.32 hr (range, 1.00-1.67 hr), and clearance half-time of 0.11±0.01 hr (range, 0.09-0.12 hr). In particular, the Cmax and AUC increased in proportion to the dose, but no dose proportionality was observed.
Efficacy of Abcertin®
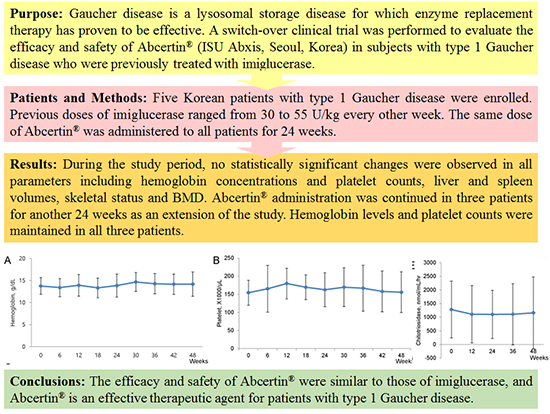
The mean hemoglobin concentration was 13.76±1.89 g/dL (range, 12.30-16.20 g/dL) at baseline, and increased to 13.86±2.61 g/dL after 24 weeks of treatment. The mean platelet count was 154.4±34.62×103/µL (range, 113-209×103/µL) at baseline and increased to 162.60±47.04×103/µL after 24 weeks of treatment. However, both of these changes were not statistically significant (Table 2, Fig. 1).
The mean ACE level increased from 79.42±31.77 U/L at baseline to 81.50±45.84 U/L after 24 weeks of drug administration. The mean acid phosphatase level decreased from 18.68±8.71 IU/L at baseline to 17.10±4.77 IU/L after 24 weeks. The mean chitotriosidase level decreased from 1,279.82±1,041.47 nM/mL/hr at baseline to 1,103.76±884.36 nM/mL/hr after 24 weeks. The mean liver volume decreased from 1.03±0.19 MN at baseline to 0.93±0.09 MN after 24 weeks. The mean spleen volume increased from 3.47±1.21 MN at baseline to 3.61±1.65 MN after 24 weeks of drug administration. However, none of these changes were statistically significant.
No changes in osteosclerosis were observed because the osteosclerosis score was 0 (none) at the baseline and 0 (none) after 24 weeks of drug administration. For osteonecrosis, one subject improved from a score of one point (mild) to zero points (none). The mean score changed from 0.20±0.45 points at baseline to 0.00±0.00 points after 24 weeks of drug administration. The mean lumbar spine Z-score increased from -1.27±0.40 at baseline to -0.80±0.53 after 24 weeks of drug administration. The mean femur neck Z-score increased from -0.43±1.37 at baseline to -0.20±1.35 at 24 weeks. However, no statistically significant differences were observed for all of these second efficacy endpoints (Table 2).
Safety
No significant changes in systolic/diastolic blood pressure, heart rate, and body temperature were found during the study period. Adverse events occurred in all five subjects, as follows: three cases of nasopharyngitis and one case of acute tonsillitis; two cases of arthralgia; one case of diarrhea and one case of abdominal pain; one case of cough; and one case of vaginal discharge. There were no serious adverse events associated with administration of the study drug. In addition, no adverse events were potentially life threatening or required withdrawal from the study. No patients generated antibodies against Abcertin® after drug administration.
DISCUSSION
This study was a phase 2 multi-center, open-label, switch-over trial to assess the safety and efficacy of Abcertin® in patients with type 1 Gaucher disease previously treated with Cerezyme®. Abcertin® was effective and well tolerated in patients with type 1 Gaucher disease. Hemoglobin concentrations and platelet counts, liver and spleen volumes, skeletal status, and BMD were maintained, and no clinically significant adverse events occurred in all patients.
The introduction of ERT has significantly impacted the treatment of type 1 Gaucher disease. At present, available enzyme therapies include imiglucerase, velaglucerase alfa, and taliglucerase alfa, all of which are generally administered intravenously every other week (6, 8, 17). Unlike imiglucerase, velaglucerase alfa has the same amino acid sequence as native human glucocerebrosidase and is produced by proprietary gene activation technology in HT-1080 human fibroblast cells (18). It is a monomeric glycoprotein (~63 kDa, containing 5 potential N-linked glycosylation sites) that targets macrophages via mannose receptors, and acts to degrade accumulated glucocerebroside within the macrophages (19). The safety and efficacy of velaglucerase alfa were previously demonstrated in a phase 1/2, open-label study in adult patients with type 1 Gaucher disease and approved by the FDA in 2010 (9). Taliglucerase alfa is a recombinant human β-glucocerebrosidase produced by carrot plant root cells, which was approved by the FDA in 2012 as a novel ERT for patients with Gaucher disease (8).
The relatively few therapeutic options and high cost of ERT represent an inherent problem for the treatment of patients with type 1 Gaucher disease (9). Global dependence on a single product, Cerezyme®, is problematic, particularly with the recent shortage of this product (20). Although velaglucerase alpha and taliglucerase alpha was approved for the long-term treatment of Gaucher disease in >40 countries, including the Unites States, European Union member states, and Israel (21), Cerezyme® still has been used worldwide. The commercially available substrate reduction agent, miglustat (Zavesca®, Actelion, Pharmaceuticals Ltd), was also used during the global shortage despite the caveat of its restricted indication to patients who are unwilling or unable to receive IV ERT (22). However, ERT is generally more effective and safer than other therapeutic options.
Therefore, a new biosimilar to imuglucerase, Abcertin®, was recently developed to potentially reduce medical costs, particularly in underprivileged countries, and to ease supply shortages of Cerezyme®. The host cell used in Abcertin® production is a mutant strain of CHO cells with an inactivated dihydrofolate reductase gene. For the development of the recombinant cell line producing Abcertin® (ISU302), host cells were transfected with an expression vector, followed by gene amplification, subcloning, and suspension culture to obtain a cell line that produces Abcertin® with a high yield. Notably, the structural, physicochemical, and biological characteristics of the Abcertin® are comparable to Cerezyme®. Phase 1 study for Abcertin® was performed to determine the safety and tolerability and pharmacokinetics in 8 healthy subjects, including placebo for 2 subjects. As a result, there were no serious adverse events and clinically significant change in vital signs, local tolerability tests, and laboratory tests (unpublished data).
During the study period, all patients maintained normal hemoglobin levels and platelet counts >100,000/µL, and all patients maintained near normal liver and spleen volumes. The findings reported herein demonstrate that adverse events associated with Abcertin® were generally mild in severity and were mostly unrelated to the therapy. No patients enrolled in this study developed antibodies against Abcertin®, and no serious adverse events were observed regardless of administration setting or duration of exposure. Furthermore, and no patients withdrew from the study as a result of adverse events. With regard to efficacy and clinical improvement in disease-specific features, the results presented herein are comparable with those of other ERTs (5, 6, 9).
One limitation of this study is that the number of patients was small and all patients were not naïve to ERT because of the rarity of this disease in Korea. Also, the study period was relatively short and there were dose differences among the study subjects. Although a small number of patients were recruited, this study is the first to investigate the safety and efficacy of Abcertin®.
In conclusion, the efficacy and safety of Abcertin® are similar to those of Cerezyme®; thus Abcertin® can be used as an alternative therapeutic agent for patients with type 1 Gaucher disease. As Cerezyme® is relatively expensive, Abcertin® is not only a useful treatment for patients with Gaucher disease but might also help to reduce costs.



 XML Download
XML Download