

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Molecular epidemiological studies have demonstrated a strong inverse relationship between the consumption of dietary isothiocyanate (ITC) and the risk of various types of malignancies, particularly lung and the gastrointestinal tract (1). The ability of cruciferous vegetables to protect against neoplastic disease has been attributed to their high glucosinolates content. One of the most commonly studied glucosinolates is glucoraphanin, which contains 4-methylsulfinylbutyl group. Myrosinase-mediated hydrolysis of glucoraphanin results in generation of sulforaphane (SFN) responsible for antioxidant, antiproliferative and anticarcinogenic properties (2).
SFN (1-isothiocyanato-4-[methylsulfinyl]-butane) is a naturally occurring ITC with promising chemopreventive activity. SFN induces cell cycle arrest and apoptosis in many types of cancer cells (3, 4) and inhibits the progression of benign tumors to malignant tumors, angiogenesis and endothelial cell functions, and the metastatic process (5). SFN also induces the phase II carcinogen detoxification enzymes such as glutathione transferases, UDP-glucuronyltransferase, NAD(P)H:quinone oxidoreductase I and heme oxygenase-1 (HO-1), thereby allowing a diverse array of electrophilic and oxidative toxicants to be eliminated or inactivated before they cause damage to critical cellular macromolecules (6). The expression of the phase II enzymes is under the control of promoter sequence known as the antioxidant response element (ARE). The activity of ARE promoter is modulated by NF-E2-related factor 2 (Nrf2), which is sequestrated by its cytoplasmic partner, Kelch-like ECH-associated protein 1 (Keap1), which promotes its ubiquitination and degradation by the proteasome (7). Modification of two crucial cysteine residues (C273 and C288) on the Keap1 by oxidation, alkylation, or arylation results in the dissociation of Keap1/Nrf2 complex and translocation of Nrf2 to the nucleus, where it binds to promoters containing the ARE sequence and activates the transcription of a series of detoxification enzymes (8). Therefore, activation of the Nrf2 pathway in cells plays a central role in enhancing the antioxidative capacity in the protection of cells and tissues from oxidative damage. In this regard, enhancement of antioxidant capacity by SFN might induce positive responses such as increases in cell survival by elevated expression of ROS-scavenging molecules. It may protect normal cells from chemotherapy and radiotherapy that involve free radical mechanisms of cytotoxicity.
Like a multitude of synthetic substances, dietary phytochemicals selectively dysregulate cellular pathways and restore apoptosis in certain tumor cells while at the same time showing low toxicity in normal cells (9). Low doses of SFN exert antioxidant properties via Nrf2-mediated gene expression and higher doses result in pro-oxidant properties (10). At high concentrations, SFN is able to increase oxidative stress in tumor cells and continue to have beneficial anti-tumor effects by exerting cytotoxic effects. These benefits rely on ROS and SFN concentration to produce either antioxidant or pro-oxidant effects. The formation of ROS in cells does not only induce oxidative stress but also leads to the development of adaptive survival responses that result in enhanced tolerance to the subsequent cytotoxicity. A number of molecules likely contribute to this protection, including thiolrich metal-binding protein, metallothionein, γ-glutamylcysteine synthetase, glutathione S-transferase, and HO-1 (11). As previously reported, Nrf2/ARE activation is required for HO-1 induction in response to oxidative/electrophile stress (12). Several signaling pathways, including phosphatidylinositol 3-kinase (PI3K), protein kinase C (PKC), and mitogen-activated protein kinase (MAPK), are implicated in HO-1 induction (13).
SFN also induces Nrf2 activation through interactions with intracellular signaling networks, including PI3K/Akt, MAPK and PKC (14). Nrf2 phosphorylation by these kinases induces the release of Nrf2 from its repressor Keap1, thereby facilitating the translocation of Nrf2 to the nucleus (15). Indeed, overexpression or activation of MAPKs and PI3K/Akt is associated with the modulation of ARE-driven gene expression via Nrf2 activation (10). Nevertheless, the mechanisms by which these signaling pathways activate Nrf2 appear to be pleiotropic and cell type-specific. Therefore, studies on the mutual interactions that occur between various protein kinases and Nrf2 in human bronchial epithelial cells may provide an efficient approach to understanding the intracellular antioxidant defense by SFN in pulmonary diseases. To explore this we utilized a human bronchial epithelial cell line, BEAS-2B, as an in vitro model for Nrf2 activation by SFN.
Based on these findings, the present study was performed to investigate positive (Nrf2/HO-1 induction) or negative (antiproliferation) response by SFN and to evaluate upstream signaling molecules involved in these processes with the aim of a better understanding the regulatory mechanism of Nrf2/HO-1 induction.
MATERIALS AND METHODS
Reagents and cell culture
Sulforaphane, N-acetylcysteine (NAC), 2', 7'-dichlorofluorescein diacetate (DCF-DA), sodium dodecyl sulfate (SDS), dimethylsuldoxide (DMSO), cycloheximide (CHX), and antibody to β-actin were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA). Ly294002 [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one], and PD98059 (2'-amino-3'-methoxyflavone) were purchased from Calbiochem (La Jolla, CA, USA). Anti-human Nrf2 antibody, HRP-tagged secondary antibodies, and enhanced chemiluminescence (ECL) kit were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-human Akt, phospho-Akt (p-Akt), Erk1/2, and phospho-Erk1/2 (p-Erk) antibodies were from Cell Signaling Technologies (Beverly, MA, USA). Antibody against HO-1 was obtained from Stressgen Biotechnologies (San Diego, CA, USA). Cell culture media and reagents were purchased from Invitrogen (Carlsbad, CA, USA). The human bronchial epithelial cell line, BEAS-2B, was obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and maintained in BEBM basal medium supplemented with 5% fetal bovine serum and BEGM SingleQuot kit (hydrocortisone, bovine pituitary extract, insulin, gentamycin sulfate, retinoic acid, transferrin, triiodothyronine, epinephrine, and human epidermal growth factor). Cells were grown to 70% confluence in a monolayer culture in this medium for 24 hr before treatment.
Cell viability assay
Cell viability was measured using the Cell Proliferation kit II (XTT) according to the manufacturer's instruction (Roche Diagnostics, Indianapolis, IN, USA). Briefly, cells were seeded in 96-well microtiter plates and then treated with SFN or other chemicals at various concentrations for the indicated times. After incubation, 50 µL of XTT labeling mixture was added to each well and incubated for an additional 4 hr. The formazan dye formed was measured spectrophotometrically at 450 nm using a Glomax multi detection system (Promega, Madison, WI, USA). The results were expressed as an absorbance value or a percentage, based on the ratio of the absorbance of treated cells to that of controls (100%).
Determination of intracellular reactive oxygen species (ROS) levels
Intracellular ROS levels were measured using DCF-DA. Briefly, cells were treated with media or SFN at different concentrations for the indicated times, after which they were loaded with 10 µM of DCF-DA for 20 min. Next, the cells were harvested and washed twice with 1 × PBS to remove all excess DCF-DA that had not penetrated into the cells. The relative fluorescence of 30,000 live cells per group was measured at excitation and emission wavelengths of 485 and 530 nm, respectively, using a multi-well fluorescence plate reader. Using untreated cells as a reference, the intracellular ROS levels were calculated by the average of three measurements and expressed as a percentage of untreated controls.
Preparation of nuclear and cytosolic protein extracts
Nuclear extracts were prepared according to the instructions of the NE-PER® nuclear and cytoplasmic extraction kit (Pierce, Rockford, IL, USA). Briefly, cells were harvested at the indicated time, and were then re-suspended in 10 vol of CER I solution, after which they were incubated in CER II solution on ice for 1 min and homogenized. Nuclei were recovered by centrifugation at 14,000 rpm for 5 min, and the supernatant was kept as the cytosolic protein extract. The nuclear fraction was extracted for 40 min on ice in NER solution. The insoluble pellet was removed by centrifugation at 14,000 rpm for 10 min. The supernatant was used as the nuclear protein extract. Nrf2 levels in nuclear and cytosolic extracts were determined by Western blotting using anti-Nrf2 antibody.
RNA interference of Nrf2
RNA interference of Nrf2 was performed using an Nrf2-specific siRNA duplex from Invitrogen (Cat# 12990). Briefly, cells were seeded in 6-well and transfected at 50% confluency with siRNA duplex (20 nM) using lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's recommendations. Cells transfected with Stealth RNAi negative control duplex (Invitrogen) were used as controls for direct comparison. After transfection, cells were treated with DMSO or SFN (10 µM) for the indicated times, after which they were processed for Western blotting.
Western blot analysis
Whole cell lysates were prepared using RIPA buffer (1 × PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 µg/mL phenylmethanesulfonylfluoride). Briefly, proteins (40 µg per lane) were separated on NuPAGE 4-12% bis-tris polyacrylamide gels (Invitrogen) and then electrophoretically transferred to Immuno-Blot PVDF membranes. The membranes were then incubated for 2 hr at room temperature with a 1:500 dilution of anti-Nrf2, anti-HO-1, anti-p-Akt, and anti-p-Erk antibodies. Next, HRP-conjugated secondary antibody was applied at a dilution of 1:5,000 and the signal was visualized using an ECL detection kit. The blots were then stripped using a stripping buffer (100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.7) and re-probed with anti-Akt, anti-Erk, and anti-β-actin antibodies as loading controls.
Statistical analysis
Statistical comparisons were made using one-way analysis of variance (ANOVA) followed by a Tukey's post hoc correction for multiple comparisons using SPSS version 17.0 (SPSS Inc., Chicago, IL, USA). Data were expressed as the mean ± SEM. Significant differences were considered with values of P < 0.05.
RESULTS
SFN inhibits cell growth by inducing a rapid increase in intracellular ROS levels
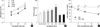
The effect of SFN on cell proliferation was examined by XTT assay after treatment with different doses (0, 5, 10 or 20 µM) of SFN for 24, 48, and 72 hr. Exposure of cell cultures to increasing concentrations of SFN led to a decrease in cell proliferation. As shown in Fig. 1A, treatment with doses of 5, 10 and 20 µM SFN resulted in a decrease in cell viability to approximately 97.7, 84.5, and 68.4% of that of control cells, respectively. A time-response experiment showed that treatment with 10 µM SFN decreased the number of living cells from approximately 84.5% of that of control cells at 24 hr treatment to 78.3% at 72 hr.
In order to investigate whether ROS can be produced under our experimental conditions, we pre-loaded BEAS-2B cells with the ROS-sensitive fluorophore DCF-DA, prior to treatment with SFN, and then measured DCF-DA fluorescence. As shown in Fig. 1B, ROS production was rapidly initiated within 10 min after SFN addition, peaked at 8 hr, and then gradually declined until 48 hr. To confirm that the SFN-induced decrease in cell viability is indeed due to its induction of ROS generation, cells were pretreated with NAC (5 mM) for 1 hr, and then exposed to SFN (0-20 µM) for 72 hr. In samples co-treated with NAC and SFN, cell viability was maintained in almost the same range as untreated controls (Fig. 1C). These results suggest that ROS play a critical role in antiproliferative response by SFN.
Erk1/2 phosphorylation is involved in SFN-induced decrease in cell viability
To determine if PI3K/Akt or MEK/Erk1/2 signaling relates to antiproliferative effect induced by SFN, we evaluated the phosphorylation of Erk1/2 and Akt by immunoblot analysis with whole cell lysates that were extracted from cells treated with SFN for the indicated time. Fig. 2A shows that SFN slightly induced Erk1/2 phosphorylation within 30 min of treatment, and then consistently increased it. However, Akt phosphorylation remained unchanged until the first 8 hr of SFN treatment, and then suppressed slightly below control levels throughout 48 hr. Effects of SFN on the phosphorylation of Erk1/2 and Akt were also observed in dose-response study. Additionally, the potential role of ROS signaling in SFN-mediated phosphorylation of AKT and Erk1/2 was evaluated using the free radical scavenger NAC. Pretreatment of cells with NAC blocked Akt phosphorylation at 2 hr or 24 hr after SFN treatment and, to a much lesser extent, Erk1/2 phosphorylation (Fig. 2B, C). We then examined cell viability during inhibition of MEK and PI3K activities using Ly294002 for PI3K and PD98059 for MEK. For this purpose, the cells were pretreated with or without inhibitors prior to treatment with different doses of SFN for 72 hr. The results indicated that pretreatment of these cells with PD98059 significantly blocked cell growth only at 10 µM SFN treatment (P < 0.05), whereas Ly294002 did not significantly affect it (Fig. 2D).
SFN increases the nuclear translocation of Nrf2 via ROS-dependent pathways
The nuclear accumulation of Nrf2 is usually considered a marker of Nrf2 activation in response to stressors. As shown in Fig. 3A, SFN treatment rapidly increased the level of Nrf2 in the nuclear fractions as expected, whereas only a weak signal of the cytoplasmic Nrf2 band was obtained. However, pretreatment of cells with NAC effectively inhibited the ability of SFN to increase Nrf2 protein levels. Similar findings were also observed in experiments using whole cell lysates. Next, we determined whether MEK/Erk1/2 and PI3K/Akt pathways were involved in SFN-induced increase in Nrf2 level at early time point (2 hr). Results showed that Nrf2 level was significantly suppressed in cells pretreated with Ly294002 and responded to some extent to PD98059 (Fig. 3B). These results imply that ROS and PI3K/Akt signalings play critical roles in induction of Nrf2 activation.
Nrf2-mediated HO-1 induction by SFN is associated with ROS and PI3K signaling
Up-regulation of HO-1 has been found to be protective in lung against oxidative stress (16) and its expression is regulated by the transcription factor Nrf2 and multiple kinase signaling pathways. To confirm whether HO-1 expression is dependent on Nrf2 activity, we employed a siRNA against Nrf2 mRNA. Cells were transfected with control siRNA or Nrf2-targeting siRNA for 48 hr. Suppression of Nrf2 expression by transfection with its siRNA completely abolished an increased in HO-1 protein induced by SFN as well as constitutive level of HO-1 (Fig. 4A). In time-response experiments, the induction of HO-1 by SFN was evident as early as 4 hr and augmentation lasted for at least 48 hr. Such effects were accompanied by an increase in Nrf2 levels (Fig. 4B).
Regarding the signaling mechanisms of HO-1 induction, several studies have suggested involvement of ROS (17). Therefore, we examined whether blockade of SFN-induced ROS generation could affect HO-1 expression at 24 hr after SFN treatment. Pretreatment with NAC inhibited an increase Nrf2 and HO-1 protein levels induced by SFN, suggesting that the effects were mediated by ROS (Fig. 5A). Next, to investigate the role of Erk1/2 and PI3K/Akt in SFN-induced HO-1 expression, we examined the effects of Ly294002 and PD98059 on SFN-induced HO-1 up-regulation. As shown in Fig. 5B, pretreatment with Ly294002 or PD98059 inhibited SFN-induced increase in HO-1 level, whereas no obvious changes were found on Nrf2 levels in cells pretreated with Ly294002. This finding suggests that PI3K/Akt signaling may regulate HO-1 expression independently Nrf2 in response to SFN.
Fig. 6 shows the time-course curves of the 5 parameters (ROS, Nrf2, HO-1, p-Akt, and p-Erk1/2) after SFN treatment. The induction levels changed with several different kinetics patterns. The levels of ROS and Nrf2 increased abruptly at early time points. This increase in ROS levels was quite rapid, peaked at 8 hr after SFN treatment, and became substantially lower and remained below control levels at 24 hr. It may be due to an increase in antioxidant capacity via Nrf2 activation. Meanwhile, Nrf2 levels increased with similar rapidity and were maintained at high levels for 48 hr. Importantly, the rapid increase in HO-1 level continued only for the first 8 hr at best. It implies that up-regulation of HO-1 expression, at least at a later period, was not solely attributable to the levels of intracellular ROS and Nrf2.
DISCUSSION
The antioxidant defense pathway is one mechanism by which the cells can respond to oxidative stress. It is also important to maintain cellular redox homeostasis and prevent the pathogenesis of many inflammatory and related diseases. A number of phytochemcals have been used to induce expression of antioxidant proteins involved in scavenging ROS (11). Nrf2 is the key transcriptional factor that serves to transmit various signals to ARE, a cis-acting regulatory element, and that plays an important role in a coordinated activation of genes encoding antioxidant proteins. In this study, we have shown the antioxidative efficacy of SFN in human bronchial epithelial BEAS-2B cells, and that upstream signaling molecules involved in these processes could play a role in maintaining Nrf2 activity. In this study, the exposure of BEAS-2B cells to SFN elicited an increase in intracellular oxidants, Erk1/2 phosphorylation, nuclear accumulation of Nrf2, up-regulation of HO-1 expression, and a decrease in cell growth. These events were effectively reversed by pretreatment with NAC, suggesting that antioxidative and antiproliferative effects induced by SFN in BEAS-2B cells are largely dependent on ROS.
Numerous studies on SFN toxicity have dealt with oxidative stress. It has been proposed that SFN-induced ROS generation is mediated indirectly by targeting the mitochondrial respiratory chain, in addition to a nonmitochondrial mechanism involving depletion in intracellular GSH concentration (18, 19). In this study, SFN treatment resulted in a significant decrease in cell viability as well as an early increase in ROS levels. Moreover, the effect of SFN on cell viability was abrogated in the presence of NAC. It provides strong evidence for the involvement of oxidative stress in antiproliferative effect induced by SFN. Some reports suggest that the activation of Erk1/2 signaling by SFN is involved in its antiproliferatve effects (20). Our study revealed that SFN strongly induce Erk1/2 phosphorylation, which is due to, at least in part, ROS generated by SFN. However, it is not clear that Erk1/2 is involved in SFN-induced antiproliferation, because it was significantly attenuated by PD98059 only at 10 µM SFN. Further studies are needed to examine the consequences of enforced activation of Erk1/2 on the effects of SFN.
The relationships between oxidative stress responses and cellular consequences are complicated due to the compensatory mechanisms regulating cellular fitness. ROS generated from endogenous and exogenous sources could influence multiple cellular processes, including signaling, proliferation, and apoptosis (21). We have shown that treatment of BEAS-2B cells with 10 µM SFN rapidly increased intracellular ROS levels, which was linked to Nrf2 activation. In addition, SFN treatment led to an increased level of Nrf2 protein in cells and such an effect was abrogated in the presence of NAC. These findings are supported by earlier reports that SFN not only requires Nrf2 activation for the induction of Phase II enzymes, but also increase the Nrf2 protein levels (22). It is possible that the activation of Nrf2 is mediated by mechanisms that lead to its stabilization as well as the regulation via its synthesis, thus increasing levels of cellular Nrf2 and subsequent transcriptional activation. Therefore, it would be rational to assume that significant increase in intracellular ROS concentrations, subsequently leading to the strong induction of HO-1 via the Nrf2 signaling, is a part of the defense system of BEAS-2B cells under oxidative stress.
In this study, because elevated ROS levels were then gradually decreased, presumably due to induction of antioxidant proteins through Nrf2/ARE activation, we originally speculated that Nrf2 activation through transient ROS generation is the part of the early response, and that other pathways including MAPKs and PI3K/Akt may be involved in its activation by SFN. However, chemical blockage of either Erk1/2 or Akt suppressed to some extent the increase of intracellular Nrf2 levels at 2 hr after SFN treatment. These findings suggest that activation of Erk1/2 or Akt at the early phase, at least in part, contributes to an increase in Nrf2 level by SFN.
Although oxidative stress can cause cell death, moderate amounts of ROS may mediate the intracellular signal transduction leading to transcriptional activation of the adaptive genes. In agreement with this notion, our present study demonstrates that alterations of the cellular redox status by SFN immediately turn on the cellular signaling cascades in such a way activating HO-1 to rescue the cells from subsequent oxidative stress. HO-1 catalyzes the degradation of pro-oxidant heme to biliverdin, carbon monoxide and ferrous ion, and has putative cytoprotective, antioxidative, and anti-inflammatory properties (23). Bronchial inflammation is a key feature of many diseases of the respiratory tract and occurs as a consequence of inhalation of allergens and environmental pollutants, microbial infection, and cigarette smoking. Oxidant injury mediates many of the toxic effects induced by environmental stressors and plays a pivotal role in the pathogenesis of pulmonary diseases (24).Therefore, roles of antioxidants and enzymes neutralizing these harmful oxidants are extremely important in determination of the intracellular antioxidant defense. Several reports have indicated that SFN, alone or in combination with other phytochemicals, induces over-production of HO-1 protein, thus exerting its anti-inflammatory activities (25, 26). In various model systems, the increase of HO-1 induction conferred protection on cells from injury and cytotoxicity induced by oxidative stress, while the abrogation of its induction accelerates cellular injuries (27). In this study, we observed that SFN is capable of strongly increasing Nrf2 and HO-1 protein levels and that their inductions are inhibited by pretreatment with NAC. Knockdown of Nrf2 by siRNA potently blocked up-regulation of HO-1 expression by SFN. Together, these findings indicate that SFN-induced increase in HO-1 levels is largely dependent on ROS/Nrf2 signaling. However, though Nrf2 levels were still maintained at high levels for the first 48 hr after SFN treatment, HO-1 levels became substantially low after the first 8 hr. This result implies that that SFN is likely to induce HO-1 expression utilizing both Nrf2-dependent and -independent pathways. In our study, inhibition of Akt phosphorylation, at least in the late time point (24 hr), partially blocked up-regulation of HO-1 expression, whereas no obvious effects were found on Nrf2 levels. This finding suggests the role of the Nrf2-independent pathway in the up-regulation of HO-1 expression in response to SFN. Similarly, there is evidence for up-regulation of HO-1 expression independently of Nrf2 in response to oleanolic acid (28). It is also reported that nerve growth factor induces the expression of HO-1 by an unknown PI3K-dependent mechanism (29). Taken together, SFN seems to regulate up-regulation of HO-1 expression via Nrf2-dependent and -independent mechanisms. Much remains to be learned about how HO-1 is regulated at the molecular level, however, change in the intracellular redox state to a more oxidized environment seems to serve as a signal that warns cells of the oxidative status, thereby triggering Nrf2/HO-1 induction as part of the defense mechanism.
In summary, we have demonstrated that exposure of the BEAS-2B cells to SFN induces antioxidative and antiproliferative effects via a ROS-mediated mechanism and at least in part, by modulation of signaling mediated by PI3K and/or MEK/Erk1/2, which result in a marked increase of Nrf2 and HO-1 protein levels.





 XML Download
XML Download