

 Citation
Citation Print
Print


INTRODUCTION
Lipid emulsion is widely used for the treatment of systemic toxicity caused by either local anesthetics, including bupivacaine, or lipid-soluble non-local anesthetic drugs [12]. Amino-amide local anesthetics, such as bupivacaine, ropivacaine, lidocaine and mepivacaine, cause vasoconstriction at a low dose and vasodilation at a high dose [34567]. Systemic toxicity induced by bupivacaine causes cardiovascular collapse [8]. Vasodilation caused by anesthetics is mediated by a decrease in either the intracellular calcium concentration ([Ca2+]i) or calcium sensitization, and bupivacaine has been reported to cause vasodilation via decreased calcium sensitization [910]. The contribution of myofilament calcium sensitization to smooth muscle contraction is associated with inhibition of myosin light chain phosphatase (MLCP), mediated by either Rho-kinase or protein kinase C (PKC) [911]. As inhibition of MLCP is mediated by phosphorylation of either the phosphorylation-dependent inhibitory protein of myosin the phosphatase (CPI-17) or myosin phosphatase target subunit (MYPT) induced by PKC or Rho-kinase, dephosphorylation of CPI-17 or MYPT via inhibition of either Rho-kinase or PKC would contribute to reduced inhibition of MLCP, leading to decreased calcium sensitization [911]. Thus, Rho-kinase and PKC are very important for the regulation of calcium sensitization-mediated contraction [11]. In support of the lipid sink theory, which states that the partitioning of a specific drug by exposure to lipid emulsion is associated with its lipid solubility, lipid emulsion reverses the severe vasodilation induced by toxic doses of local anesthetics, including bupivacaine, levobupivacaine, ropivacaine, lidocaine and mepivacaine, in a lipid solubility-dependent manner [137]. In addition, lipid emulsion attenuates the vasodilation induced by a toxic level of bupivacaine by decreasing bupivacaine-induced MYPT dephosphorylation, suggesting that lipid emulsion inhibits bupivacaine-induced attenuation of calcium sensitization mediated by Rho-kinase [12]. In particular, PKC is very important for the regulation of vasoconstriction mediated by either inhibition of the catalytic subunit of MLCP via CPI-17 or caldesmon phosphorylation [911]. However, whether vasodilation induced by a toxic dose of local anesthetic is mediated by a decrease in PKC-induced calcium sensitization-mediated vasoconstriction remains unknown. In addition, the effect of lipid emulsion on the vasodilation caused by a toxic dose of local anesthetic during PKC-induced contraction in association with regulation of calcium sensitization also remains unknown. Therefore, the goal of this in vitro study was to investigate the effect of lipid emulsion on vasodilation caused by toxic doses of bupivacaine and mepivacaine during contraction induced by PKC activator phorbol 12,13-dibutyrate (PDBu)-mediated calcium sensitization in an isolated endothelium-denuded rat aorta and to determine the associated cellular mechanism. Based on previous reports, we tested the hypothesis that lipid emulsion would greatly inhibit vasodilation induced by a toxic dose of bupivacaine compared with that induced by a toxic dose of mepivacaine [137].
MATERIALS AND METHODS
All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee of Gyeongsang National University. All experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals prepared by the Institute for Laboratory Animal Research.
1. Preparation of aortic rings for tension measurements
Preparation of aortic rings for tension measurements was performed as previously described [12]. Male Sprague-Dawley rats weighing 250–300 g were anesthetized by an intramuscular injection of Zoletil 50 (15 mg/kg, Virbac Laboratories, Carros, France). The descending thoracic aorta was dissected free from surrounding connective tissues and fat under microscopic guidance in a Krebs solution bath consisting of 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.4 mM CaCl2, 25 mM NaHCO3, and 11 mM glucose. The endothelium was removed from all aortic rings by inserting a 25-gauge needle into the lumens of the rings and gently rubbing the rings for a few seconds. The aorta was suspended on a Grass isometric transducer (FT-03, Grass Instrument, Quincy, MA, USA) under 3.0-g resting tension in a bath containing 10 ml Krebs solution at 37℃ and aerated continuously with 95% O2 and 5% CO2 to maintain the pH between 7.35–7.45. The rings were equilibrated at 3.0-g resting tension for 120 min, and the bath solution was changed every 30 min. After contraction induced by 10-8 M phenylephrine had stabilized, endothelial denudation of the aortic rings was confirmed by observation of less than 10% relaxation in response to acetylcholine (10-5 M). The phenylephrine was washed out of the organ bath, and isometric tension was allowed to return to the baseline. Then, the main experiments were performed according to the experimental protocols described below. Each ring was used for generation of only one concentration-response curve induced by bupivacaine, mepivacaine, or PDBu. Because local anesthetic-induced nitric oxide release attenuates vascular responses caused by local anesthetics in the endothelium-intact aorta, the Krebs solution contained the nitric oxide synthase inhibitor NW-nitro-L-arginine methyl ester (L-NAME, 10-4 M) to prevent the release of endogenous nitric oxide from the residual endothelium [4613].
2. Experimental protocols
The effect of Intralipid® (lipid emulsion) on vasodilation induced by bupivacaine and mepivacaine in an aorta precontracted with a PKC activator, PDBu, was investigated. The aortic rings were treated with lipid emulsion (0.35 and 0.8%) for 20 m before addition of PDBu (10-6 M). After PDBu (10-6 M) had produced a stable and sustained contraction in the presence or absence of lipid emulsion for 10 m, incremental concentrations of bupivacaine (10-6 to 10-3 M) or mepivacaine (10-5 to 3 × 10-3 M) were cumulatively added to the organ bath to generate concentration-response curves induced by bupivacaine or mepivacaine. In addition, the effects of a PKC inhibitor, GF109203X, or lipid emulsion on PDBu (10-6 M)-induced contraction were investigated. After PDBu (10-6 M) had produced a stable and sustained contraction in the endothelium-denuded aorta, 10-6, 3 × 10-6, or 10-5 M GF109203X was added to the organ bath, and tension was continuously monitored for 60 m. After PDBu (10-6 M) had produced a stable and sustained contraction in the endothelium-denuded aorta for 10 m, lipid emulsion (0.35 and 0.8%) was cumulatively added to examine its effect on PDBu (10-6 M)-induced contraction.
3. Fura-2 loading and simultaneous measurements of [Ca2+]i and tension
[Ca2+]i and tension were simultaneously measured as described previously [12]. Muscle strips were exposed to the acetoxymethyl ester of fura-2 (fura-2/AM, 10 µM) in the presence of 0.02% cremophor EL for 5–6 h at room temperature. After loading, a muscle strip was washed with Krebs solution at 37℃ for 20 m to remove any uncleaved fura-2/AM and held horizontally in a temperature-controlled, 7-ml organ bath. One end of the muscle strip was connected to a force-displacement transducer (MLT050, AD Instruments, Colorado Springs, CO, USA) to monitor muscle contraction. The muscle strip was illuminated alternatively (48 Hz) at two excitation wavelengths (340 and 380 nm). Fluorescence intensity at 500 nm (F340 and F380) was measured with a fluorometer (CAF-110, Jasco, Tokyo, Japan). The ratio of F340 to F380 (F340/F380) was calculated as an indicator of [Ca2+]i. Therefore, the F340/F380 ratio and tension obtained from 10-6 M PDBu-stimulated aortic strips were taken as 100 and 100%, respectively. Isometric contractions and the F340/F380 ratio were recorded with a PowerLab/400 using the chart program (AD instruments). The muscle strips were placed under an initial resting tension of 3.0 g. All strips obtained from the same animal were used in different experimental protocols. When both [Ca2+]i and contraction induced by 10-6 M PDBu reached steady-state levels, incremental concentrations of bupivacaine (10-6 to 10-3 M) were added cumulatively.
4. Cell culture
Vascular smooth muscle cells (VSMCs) were isolated from the thoracic aortas of male rats by enzymatic dissociation and grown in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin as described previously [12]. Cells were subcultured twice per week by harvesting with trypsin/ethylenediaminetetraacetic acid and seeding into flasks at a density of 7.5 × 105/mm2. For the experiments, cells between passage numbers 2 and 10 were seeded into dishes (107/100-mm dishes), fed every other day, and used at confluence (6–7 days). Cells were deprived of serum overnight prior to treatment.
5. Western blot analysis
Western blot analysis was performed according to the method described by Lee et al. [14]. Cells were lysed in an RIPA buffer (Cell Signaling Technology, Beverly, MA, USA) to obtain total cell lysates, which were centrifuged at 14,000 rpm for 15 min at 4℃. The protein concentrations were determined using the Bradford method. For sample loading, equal volumes of 2× sodium dodecyl sulfate sample buffer (0.1 mol/L Tris-HCI, 20% glycerol, 4% sodium dodecyl sulfate, and 0.01% bromophenol blue) and supernatant fractions from the lysates were mixed. Proteins (30 µg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis for 90 min at 110 V. The separated proteins were transferred to polyvinylidene difluoride membranes for 20 min at 20 mA using SD Semi-dry Transfer Cells (Bio-Rad Laboratories, Hercules, CA, USA). After blocking of the membranes using 5% bovine serum albumin (BioShop, Burlington, Canada) for phospho-PKC and 5% skim milk (Becton, Dickinson and Company, Maryland, USA) for PKC, phospho-CPI-17, and CPI-17 in Tris-buffed saline (pH 7.0), the membranes were incubated overnight at 4℃ with specific antibodies (ant-phospho-PKC, anti-PKC, anti-phospho-CPI-17, and anti-CPI-17 antibodies) diluted 1:1000 in 5% bovine serum albumin or 5% skim milk in Tris-buffed saline containing Tween-20. Bound antibody was detected with horseradish peroxidase-conjugated anti-rabbit IgG. The membranes were washed and developed using a western blotting luminol reagent system (iNtRON Biotechnology, Houston, TX, USA) and autoradiography.
6. Materials
All drugs were of the highest purity commercially available. PDBu, GF109203X, L-NAME and phenylephrine were obtained from Sigma Aldrich (St. Louis, MO, USA). Fura-2 was purchased from Molecular Probes (Eugene, OR, USA). Intralipid® 20% was acquired from Fresenius Kabi Korea (Seoul, Korea). Bupivacaine was obtained from Reyon Pharmaceutical Co., Ltd. (Seoul, Korea). Anti-PKC and anti-phospho-PKC (pan) antibodies were purchased from Cell Signaling Technology. Anti-CPI-17 and anti-phospho-CPI-17 (Thr 38) were acquired from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All concentrations were expressed as the final molar concentration. PDBu, GF109203X and Fura-2/AM were dissolved in dimethyl sulfoxide. Unless stated otherwise, all drugs were dissolved in distilled water.
7. Data analysis
The values are presented as the mean ± SD. Vasodilation induced by bupivacaine, mepivacaine and GF109203X is expressed as a percentage of the maximal contraction induced by PDBu. The effect of lipid emulsion on vasodilation induced by bupivacaine or mepivacaine was analyzed by a two-way repeated measures analysis of variance (ANOVA), followed by Bonferroni's post-test (Prism 5.0, GraphPad Software, San Diego, CA, USA). The effect of GF109203X on PDBu-induced contraction was analyzed by two-way repeated measures ANOVA, followed by Bonferroni's post-test. In addition, the effects of bupivacaine on contraction and the increase [Ca2+]i induced by PDBu were analyzed by two-way repeated measures ANOVA, followed by Bonferroni's post-test. Finally, the effects of GF109203X, bupivacaine, and combined treatment with lipid emulsion and bupivacaine on PDBu-induced PKC and CPI-17 phosphorylation in rat aortic VSMCs were analyzed by one-way ANOVA, followed by Bonferroni's post-test. N indicates the number of isolated aortic rings or the number of rats from which thoracic aortic rings or strips were obtained in tension measurements or simultaneous measurements of [Ca2+]i and tension, and N indicates the number of independent experiments in western blot analysis.
RESULTS
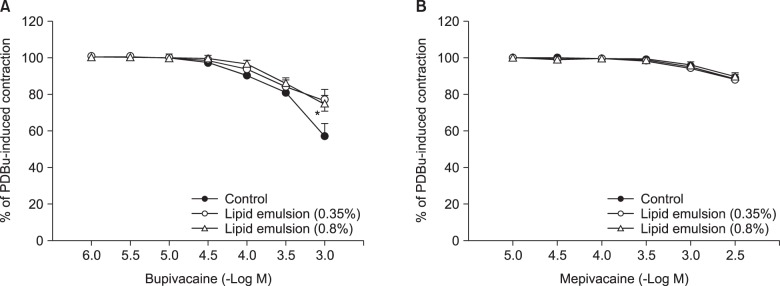
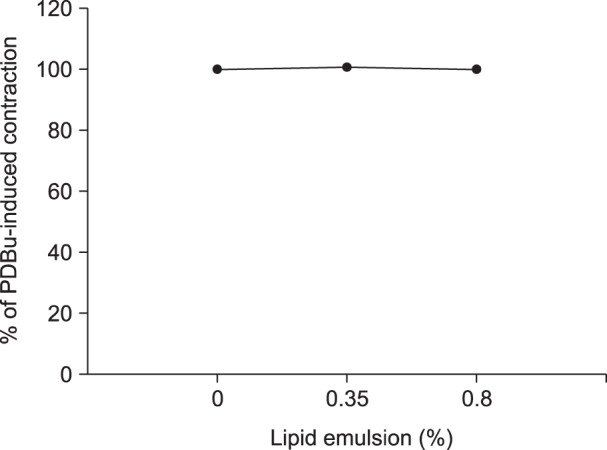
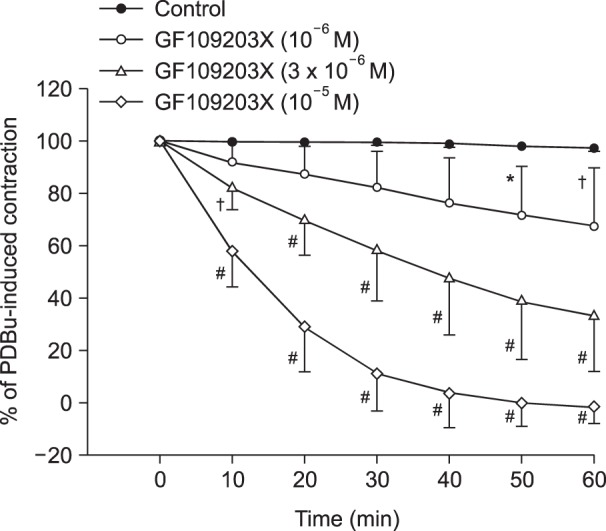
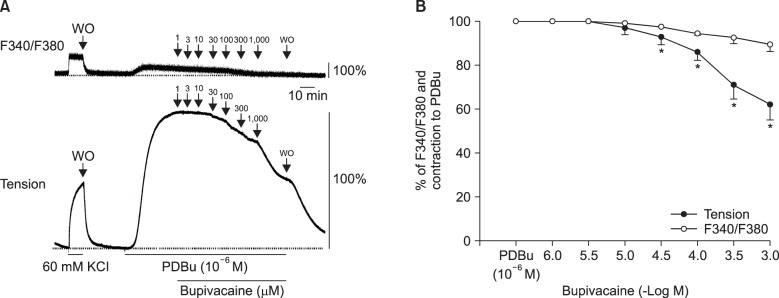
Lipid emulsion (0.35 and 0.8%) attenuated bupivacaine-induced maximal vasodilation (Fig. 1A; P < 0.001 versus control). However, it had no effect on mepivacaine-induced vasodilation (Fig. 1B). Lipid emulsion itself had no effect on PDBu-induced contraction (10-6 M) (Fig. 2). GF109203X (10-6, 3 × 10-6, or 10-5 M) attenuated PDBu-induced vasoconstriction (Fig. 3; P < 0.05 versus the control at 50 and 60 m). The magnitude of bupivacaine-induced vasodilation (3 × 10-5 to 10-3 M) was higher than that of the bupivacaine-induced [Ca2+]i decrease (Fig. 4; P < 0.001 versus control).
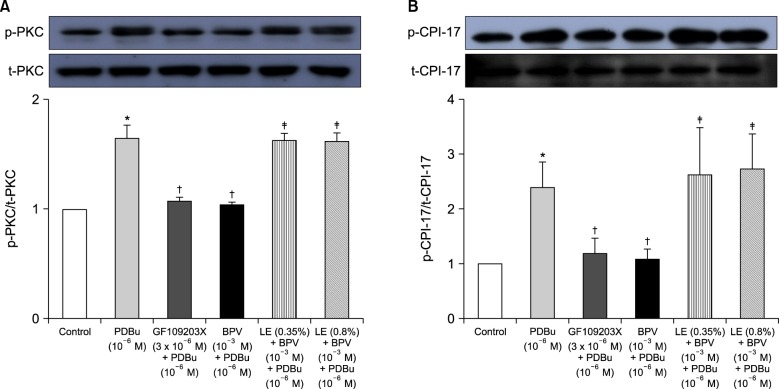
PDBu promoted PKC and CPI-17 phosphorylation (Fig. 5A and B; P < 0.001 versus control). Furthermore, bupivacaine (10-3 M) and GF109203X (3 × 10-6 M) attenuated PDBu (10-6 M)-induced PKC phosphorylation (Fig. 5A; P < 0.001 versus PDBu alone), whereas lipid emulsion (0.35 and 0.8%) attenuated bupivacaine-mediated inhibition of PDBu (10-6 M)-induced PKC phosphorylation (Fig. 5A; P < 0.001 versus combined treatment with bupivacaine and PDBu). Bupivacaine (10-3 M) and GF109203X (3 × 10-6 M) attenuated PDBu (10-6 M)-induced CPI-17 phosphorylation (Fig. 5B; P < 0.001 versus PDBu alone), whereas lipid emulsion (0.35 and 0.8%) attenuated bupivacaine-mediated inhibition of PDBu (10-6 M)-induced CPI-17 phosphorylation (Fig. 5B; P < 0.001 versus combined treatment with bupivacaine and PDBu).
DISCUSSION
This is the first study to suggest that lipid emulsion attenuates the vasodilation induced by a toxic dose of bupivacaine via inhibition of bupivacaine-induced PKC and CPI-17 dephosphorylation. The major findings of this in vitro study are as follows: 1) lipid emulsion attenuated bupivacaine-induced vasodilation in an endothelium-denuded aorta precontracted with the PKC activator PDBu but had no effect on mepivacaine-induced vasodilation; 2) the magnitude of bupivacaine-induced vasodilation was higher than that of the bupivacaine-induced [Ca2+]i decrease; and 3) lipid emulsion attenuated bupivacaine-mediated inhibition of PDBu-induced PKC and CPI-17 phosphorylation.
Bupivacaine causes vasodilation via decreased calcium sensitization in isolated endothelium-denuded rat aortas precontracted with phenylephrine [10]. Bupivacaine-induced vasodilation involves inhibition of MYPT phosphorylation induced by the Rho-kinase activator NaF, suggesting that bupivacaine has an inhibitory effect on Rho-kinase-mediated calcium sensitization [12]. Vascular smooth muscle contraction is mediated by an increased [Ca2+]i or by myofilament calcium sensitization [9]. Calcium sensitization is associated with steeper slopes of calcium-tension curves attributed to a vasoconstrictor than those attributed to a high KCl concentration [15]. In other words, calcium sensitization implies that the magnitude of increase in vasoconstrictor-induced tension is higher than that of the vasoconstrictor-induced [Ca2+]i increase [15]. Calcium sensitization is mediated by inhibition of MLCP through phosphorylation of either CPI-17 or MYPT induced by PKC and Rho-kinase [911]. The PKC inhibitor GF109203X decreased PDBu-induced vasoconstriction (Fig. 3), suggesting that this vasoconstriction is mediated by PKC. In an aorta precontracted with the PKC activator PDBu, the magnitude of the reduction in tension caused by bupivacaine (3 × 10-5 to 10-3 M) was higher than that of the decreased [Ca2+]i (Fig. 4B), suggesting that a toxic dose of bupivacaine causes decreased calcium sensitization through inhibition of the PKC-mediated pathway. In addition, the PKC activator PDBu promoted PKC and CPI-17 phosphorylation, whereas the PKC inhibitor GF109203X and bupivacaine (10-3 M) attenuated PDBu-induced PKC and CPI-17 phosphorylation (Fig. 5). As CPI-17 is a downstream effector activated by PKC phosphorylation, which is induced by vasoconstrictors and results in calcium sensitization, these results suggest that bupivacaine causes vasodilation via an inhibitory effect on PKC-mediated CPI-17 phosphorylation, leading to decreased calcium sensitization [91116]. The magnitude of bupivacaine (10-3 M)-induced maximal vasodilation has been reported to be 93 ± 3% of the maximal contraction induced by the Rho-kinase activator NaF in an endothelium-denuded aorta [12]. However, in the current study, the magnitude of bupivacaine (10-3 M)-induced maximal vasodilation was 44 ± 7% of the maximal contraction induced by the PKC activator PDBu. Rho-kinase that has been activated by a vasoconstrictor causes calcium sensitization via CPI-17 and/or MYPT phosphorylation, leading to increased calcium sensitization through enhanced inhibition of MLCP compared with PKC phosphorylation [91116171819]. In contrast, PKC phosphorylation induced by a vasoconstrictor causes calcium sensitization via CPI-17 phosphorylation [91116171819]. Thus, Rho-kinase has a relatively greater contribution to calcium sensitization than PKC because inhibition of MLCP by Rho-kinase and PKC is mediated by CPI-17 and/or MYPT or CPI-17 alone, respectively [91116171819]. In other words, the relative decrease in bupivacaine-induced vasodilation in the aorta precontracted with the PKC activator PDBu in the current study compared with the aorta precontracted with the Rho-kinase stimulant NaF in the previous study may be ascribed to a relatively lower contribution of PKC to calcium sensitization-mediated contraction compared with Rho-kinase [12]. Further study of the cellular signaling pathway upstream from bupivacaine-mediated inhibition of PKC and CPI-17 phosphorylation evoked by PDBu is needed to elucidate the detailed signaling pathway.
Lipid emulsion is effective for the treatment of toxicity induced by local anesthetics and lipid-soluble non-local anesthetic drugs [12]. The mechanism underlying the effects of lipid emulsion treatment in systemic toxicity induced by local anesthetics or other drugs involves a lipid sink, an enhanced fatty acid supply, a cardiotonic effect, inhibition of the blockade of sodium channels induced by local anesthetics, Akt-induced glycogen synthase kinase-3β phosphorylation and drug redistribution [1202122]. Pre-treatment or post-treatment with lipid emulsion in in vitro studies performed using isolated vessels has been demonstrated to reverse or attenuate the vasodilation induced by toxic doses of local anesthetics in a lipid solubility-dependent manner [371223]. In H9c2 cells, lipid emulsion-mediated inhibition of apoptosis induced by a toxic dose of bupivacaine is enhanced compared with that induced by a toxic dose of mepivacaine [24]. In addition, pre-treatment or post-treatment with lipid emulsion has been shown in in vivo studies to be effective during recovery from cardiac arrest induced by bupivacaine but to have no effect on recovery from cardiac arrest induced by mepivacaine, suggesting that the efficacy of lipid emulsion treatment is partly dependent on the lipid solubility of the local anesthetic [2526]. All of these previous studies indicate that lipid emulsion-mediated recovery from cardiovascular collapse due to local anesthetic toxicity can be partly attributed to lipid emulsion-mediated sequestration of the local anesthetics, which is dependent on their lipid solubility [1371223242526]. Similar to previous reports, in the current study, lipid emulsion attenuated vasodilation induced by a toxic dose of bupivacaine (Fig. 1A) but had no effect on that induced by mepivacaine (Fig. 1B), suggesting that lipid emulsion-mediated attenuation of vasodilation is dependent on the lipid solubility of the local anesthetic (h-octanol/buffer partition coefficient: bupivacaine [346] versus mepivacaine [21]) [37122324252627]. As lipid emulsion attenuated bupivacaine-mediated inhibition of PDBu-induced PKC and CPI-17 phosphorylation (Fig. 5), the results suggest that lipid emulsion-mediated attenuation of bupivacaine-induced vasodilation involves an inhibitory effect of lipid emulsion on bupivacaine-induced PKC and CPI-17 dephosphorylation. In other words, considering the results of the current tension study and those of previous reports, lipid emulsion-mediated sequestration of bupivacaine, a local anesthetic with relatively high lipid solubility, appears to partly contribute to inhibition of bupivacaine-induced PKC and CPI-17 dephosphorylation [371223242526]. In addition, lipid emulsion increases [Ca2+]i [28]. As the conventional PKC α and β isoforms are calcium-dependent isoenzymes, the increased [Ca2+]i induced by lipid emulsion may contribute to attenuation of bupivacaine-induced vasodilation through enhancement of PKC activation during PDBu-induced contraction [2829]. However, as lipid emulsion itself had no effect on PDBu-induced contraction (Fig. 2), lipid emulsion-mediated attenuation of bupivacaine-induced vasodilation was not associated with lipid emulsion-mediated enhancement of PDBu-induced contraction.
Systemic toxicity induced by bupivacaine results in cardiovascular collapse [8]. Lipid emulsion-mediated attenuation of severe vasodilation induced by a toxic dose of bupivacaine, which promotes dephosphorylation of PKC and CPI-17, may have a beneficial effect on vascular tone recovery from severe vascular collapse caused by bupivacaine toxicity. This effect appears to be associated with the lipid solubility of local anesthetics. The results of current and previous studies suggest that bupivacaine toxicity would be more responsive to lipid emulsion treatment than mepivacaine toxicity [1372324252627]. In addition, lipid emulsion is effective for the treatment of seizures, which is considered one of central nervous system symptoms of local anesthetic systemic toxicity that occurs before cardiovascular collapse, supporting the lipid sink theory because the lipid emulsion itself does not affect brain metabolism. Further, the American Society of Regional Anesthesia and Pain Medicine also recommends the early use of lipid emulsion for the treatment of local anesthetic systemic toxicity [13031]. Thus, considering the current results supporting the lipid sink theory, lipid emulsion treatment at the early stage of local anesthetic systemic toxicity appears to be optimal for improving the prognosis of patients suffering from bupivacaine-induced systemic toxicity. However, the clinical relevance of lipid emulsion-mediated attenuation of vasodilation induced by a toxic dose (10-3 M) exceeding the toxic plasma concentration (10-5 M) of bupivacaine, causing hypotension due to systemic toxicity, could be modified by the following factors [32]. First, the vascular response (vasoconstriction at a low dose and vasodilation at a high dose) induced by local anesthetics is attenuated by endothelial nitric oxide release, and lipid emulsion itself increases left ventricular systolic pressure via blockade of nitric oxide release [461333]. These two factors may antagonize each other [461333]. However, we used an endothelium-denuded rat aorta in the current study. Second, organ blood flow and total peripheral vascular resistance are mainly determined by small resistance arterioles, whereas we used a rat thoracic aorta, which is regarded as a conduit vessel, in the current study [34].
In conclusion, these results of this study suggest that lipid emulsion attenuates the vasodilation induced by a toxic dose of bupivacaine via inhibition of bupivacaine-induced PKC and CPI-17 dephosphorylation in an isolated endothelium-denuded rat aorta precontracted with PDBu, which may be associated with the relatively high lipid solubility of bupivacaine.
 XML Download
XML Download