

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Abnormalities involving the proximal segment of the long arm of chromosome 14 (14q) are rare. Clinical phenotypes of duplications on 14q vary, with autistic disorder, mental retardation, delayed development, and dysmorphic features being the common phenotypes [123]. Several studies have attempted to elucidate the correlation between these clinical phenotypes and candidate genes. However, because of insufficient reports and genetic studies, establishing a correlation between genotype and the corresponding phenotype has been difficult. In this study, we report a newborn with an 18.3 Mb large duplication in the 14q11.2q21.1 that was identified by a chromosomal microarray.
Our case was a 1-month-old newborn girl born at 41 weeks of gestation through vaginal delivery. She was the third child, and her parents were both 33 yr old. Her mother had an obstetric history of gravida 4, para 3, and abortus 1. The first and second children did not have developmental problems or intellectual disabilities, and there was no family history. The birth weight, height, and head circumference of the patient were within normal ranges (2,940 g [3rd-10th percentile], 47.5 cm [10th-25th percentile], and 32.5 cm [10th-25th percentile], respectively). Cardiac murmur was detected, and hypoxemia (SpO2, 68-85%) was observed after birth. Echocardiography indicated a ventricular septal defect, pulmonary stenosis, and an overriding aorta, correlating to tetralogy of Fallot (TOF). The patient had bilateral clubfoot. However, other dysmorphic features in the extremities and face were unnoticeable.
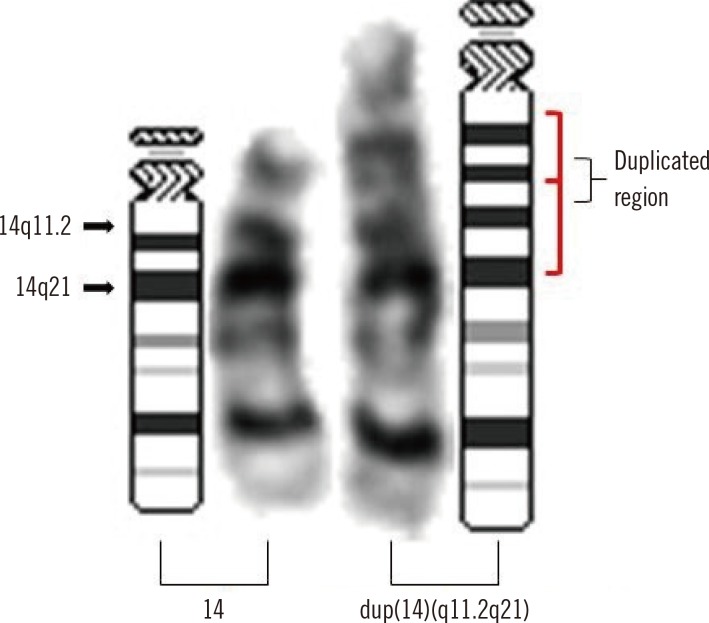
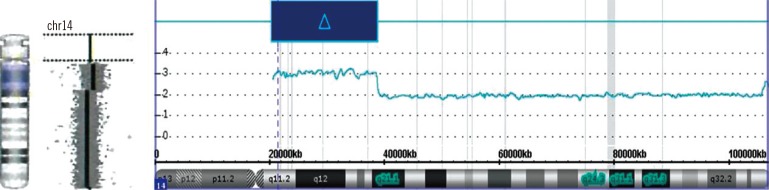
G-banding chromosome analysis of phytohemagglutinin (PHA) stimulation on cultured lymphocytes from peripheral blood showed an insertion in the proximal segment of 14q in all 25 metaphase cells (Fig. 1). At the level of 550-band resolution, the breakpoint and origin of the inserted fragment could not be clearly identified. We further investigated copy number alterations using Cytoscan 750K array (Affymetrix, Santa Clara, CA, USA). An approximately 18.3-Mb gain from 14q11.2 to 14q21.1 (chr14:20,516,277-38,826,881) was identified (Fig. 2). The karyotype was 46,XX,dup(14)(q11.2q21).arr 14q11.2q21.1 (20,516,277-38,826,881)x3 according to the International System for Human Cytogenetic Nomenclature (2013). Chromosome analyses in both parents showed normal karyotypes, indicating a de novo duplication mechanism.
Compared with other patients with 14q duplications, our patient showed multiple cardiac anomalies. We found 47 duplication cases in the 14q11.2q21.1 region from DECIPHER (DatabasE of Chromosomal Imbalance and Phenotype in Human using Ensemble Resources, http://www.decipher.sanger.ac.uk). The most frequent phenotypes were intellectual disability (34.0%) and autism (19.1%); only two patients showed cardiac anomalies. One patient (DECIPHER ID: 283016) with a 0.39-Mb duplication in 14q12 (chr14:29,829,325-30,220,298) had autistic spectrum disorder, malformation of the heart and great vessels, nystagmus, and TOF. The other patient (DECIPHER ID: 287854) with a 0.37-Mb duplication in 14q11.2 (chr14:20,424, 580-20,796,696) had a hypoplastic right heart and malformation in the heart and great vessels.
The 18.3-Mb duplication observed in our patient was large enough to be detected by chromosomal analysis. This region contains 446 genes and is associated with 32 phenotypes (NCBI database: http://www.ncbi.nlm.nih.gov/mapview/maps.cgi? [Build 35]). The only cardiac anomaly-associated gene in this region is myosin heavy chain 6 (MYH6) [4]. However, a missense mutation in MYH6, not a duplication, is known to be associated with cardiac anomalies. Most congenital heart defects arise from abnormal heart development in embryogenesis. During heart development, the dose of a gene product available at a specific time and location is important for normal cardiogenesis [5]. Hence, the cardiac anomalies observed in our patient may have resulted from dosage imbalances during cardiac development. Monfort et al. [2] reported that quantitative trait loci (QTLs) in 14q11.2 controlled stature. Similarly, we propose that the duplication observed in our patient supported the presence of a putative QTL in 14q that controlled heart development. The current results indicated that a duplication in chromosome 14 could lead to congenital heart defects.
Clubfoot can be associated with chromosomal abnormalities [67]. Six patients from DECIPHER (ID: 1366, 248559, 249644, 249887, 277167, and 279247) have anomalies of the extremities and overlapping duplicated regions with our case. The critical region for anomalies of extremities is still uncertain, because there was no common region among these cases.
Two patients (nssv578665, nssv578667) with 19-Mb duplications overlapping with the 14q11.2q21.1 region have been reported in the International Standards for Cytogenomic Arrays database (http://www.ncbi.nlm.nih.gov/dbvar/). Both had facial dysmorphism, hypertonia, intellectual disability, and developmental delay. Because of the young age of our patient, intellectual disability and delayed development could not be evaluated. Follow-up examinations for expected phenotypes are necessary.
Thus, we confirmed an insertion of unknown origin in a congenital heart defect case using chromosomal microarray analysis. Chromosomal microarray is useful for determining the size and precise breakpoints of abnormalities, regardless of whether they could be detected by G-banding analysis.
 XML Download
XML Download