

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Metachromatic leukodystrophy (MLD; MIM# 250100) is suspected when leukocyte arylsulfatase A (ARSA; E.C. 3.1.6.8) activity is much lower than that in normal individuals. However, this cannot be used to distinguish between patients with MLD and those with ARSA pseudodeficiency (PD), in whom ARSA activity is in the same range as that in MLD patients but who do not show any clinical symptoms of the disease. In such cases, MLD can be confirmed by determining mutations in ARSA (MIM# 607574) [1, 2, 3].
Approximately 189 ARSA mutations have been reported in Human Gene Mutation Database (HGMD) thus far [4]. Three types of ARSA alleles decrease ARSA activity: (1) disease-causing ARSA-MLD allele in a homozygous or compound heterozygous state; (2) ARSA-PD allele, which results in below-average ARSA activity; and (3) combination ARSA-MLD and ARSA-PD alleles, in which the ARSA-MLD allele is present in a cis configuration with the ARSA-PD allele [5, 6]. The most common disease-causing mutations include c.465+1G>A, pIle181Ser, c.1210+1G>A, and p.Pro428Leu in Europeans and c.302G>A in the Japanese population [6, 7, 8]. In contrast, only a few genetic mutations, such as c.1107+1G>T and c.302G>A, have been reported in Koreans thus far [9, 10, 11].
To distinguish patients with MLD from those with PD or from heterozygotes, we performed genetic analysis of DNA obtained from dried blood spots (DBSs) of Korean patients with suspected MLD who underwent an analysis of ARSA activity in leukocytes. We then identified mutations in ARSA by comparing ARSA activity and sulfatide levels.
DBSs and leukocytes were collected from the peripheral blood of seven subjects, whose ARSA activities were analyzed at the Seoul National University Bundang Hospital, Korea. Of these seven subjects, three were children with clinical symptoms of late infantile MLD, two were adults with MLD-like symptoms, and two were obligate heterozygous adults who were parents of patient 1 (Table 1). Patients 1 and 3 showed gait disturbances. Radiological analysis demonstrated that these patients showed progression of white matter disease in the brain. Informed consent was obtained from all adult patients and from parents of children with suspected MLD for mutational analysis. Residual DBSs stored at -70℃ after examining the enzyme activity were used for mutational analysis. EDTA-anticoagulated blood was collected from the parents of patient 1. This research was approved by the Institutional Review Board of the Seoul National University Bundang Hospital (No. B-1308-216-012).
ARSA activity in the peripheral blood leukocytes was assayed by using an artificial substrate, p-nitrocatechol sulfate. Results were compared with those of normal controls and were expressed in nmol/min/mg protein [3]. Sulfatide concentrations were determined by using procedures developed in our previous study [12]. In brief, sulfatides were eluted from DBSs, and working solutions were prepared, including controls and internal standard. The sulfatides eluted were analyzed and were quantified by using ultra-performance liquid chromatography and tandem mass spectrometry.
DNA was extracted from DBSs by using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. Primers specific to 8 exons of ARSA were used to amplify the exons and flanking intronic regions. PCR was performed by using GeneAmp PCR System 9700 Thermal Cycler (Applied Biosystems, Foster City, CA, USA). Direct sequencing was performed by using ABI 3730xl DNA Analyzer (Applied Biosystems) with BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). HGMD Professional database was used to verify novel mutations identified (Reference sequence: NM_000487.5). Sequencher version 5.0 (Gene Codes Corporation, Ann Arbor, MI, USA) and Alamut version 2.0 (Interactive Biosoftware, Rouen, France) were used for mutational analysis.
RNA was extracted from the specimen with the novel mutation by using TRIzol Reagent (Life Technologies, Carlsbad, CA, USA), and cDNA was generated by using SuperScript III Reverse Transcriptase (Life Technologies), according to the manufacturer's instructions. Reverse transcription-PCR-based sequencing was performed by using a set of primers (forward: 5'-GCCATA GGGGACCTGGGGCT-3', reverse: 5'-TGAAGCTGTTTCAGGGCTTGCAG-3'). Direct sequencing of the PCR product was performed as described above. Mutation nomenclature used in this study follows the recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/) [13].
Results of ARSA activity, sulfatides levels, and mutational analysis are summarized in Table 1. ARSA activity in the seven individuals ranged from 0.08 to 0.47 nmoL/min/mg protein, which was lower than the normal range. ARSA activity in patients 1-3 was much lower than that in normal controls, suggesting that these patients had ARSA deficiency. Total sulfatide concentration was markedly increased in three patients with MLD compared with that in individuals with MLD-PD and PD and in obligate heterozygotes, and was inversely correlated with ARSA activity by Spearman correlation analysis (Spearman's coefficient of rank correlation, ρ=0.929, P=0.0025).
Direct sequencing of ARSA by using DNA extracted from DBS identified several known mutation alleles of MLD, including c.256C>T, c.302G>A, and c.465+1G>A. Direct sequencing also identified variant alleles suggestive of PD, including c.1178C>G and c.1055A>G, and a novel pathogenic variant allele, c.1107+1delG, that was not previously reported in the HGMD database.
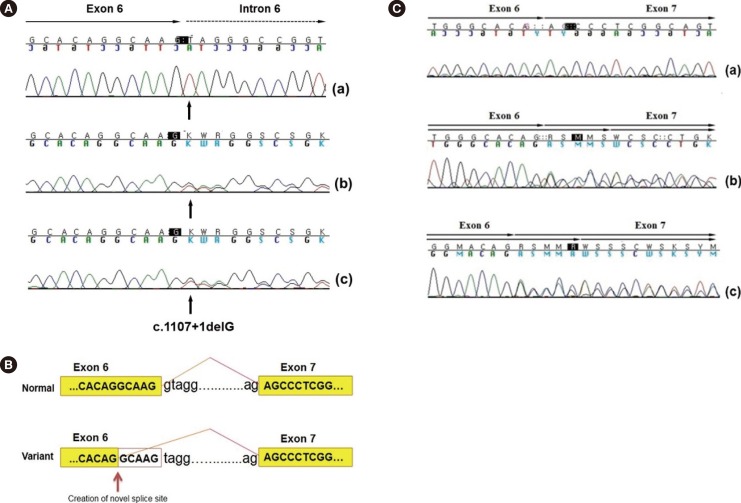
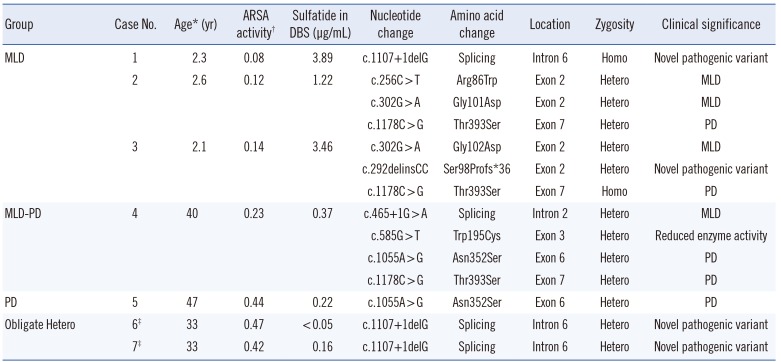
Genetic sequencing of ARSA from patient 1 identified a novel homozygous mutation, c.1107+1delG, in the splicing donor site of intron 6. Targeted mutational analysis of the patient's parents showed that they were heterozygous for the c.1107+1delG allele at the ARSA locus, suggesting an autosomal recessive inheritance pattern (Fig. 1A). The effect of nucleotide deletion on splicing was analyzed by using the cDNAs of patient 1 and her parents. The cDNAs of the parents with the heterozygous c.1107+1delG allele showed overlapping peaks surrounding the splice junction between exons 6 and 7. These overlapping peaks were not observed in the cDNA of the patient, indicating that the c.1107+1delG allele was a very strong pathogenic variant (Fig. 1B, C), according to the recent standards and guidelines for the interpretation of sequence variants [14]. Next, 90 healthy individuals without any clinical symptoms of MLD were screened to determine whether the c.1107+1delG mutation was a common nucleotide polymorphism. Mutational analysis of these individuals yielded negative results, suggesting that the c.1107 +1delG mutation may be the cause of ARSA deficiency.
MLD is caused by a deficiency of ARSA. In this study, we confirmed three patients with MLD who had homozygous or compound heterozygous ARSA-MLD alleles and identified a novel pathogenic variant allele c.1107+1delG in one of these patients. Most patients who were confirmed as having MLD showed low ARSA activity in the range that is generally associated with the disease. Cases with moderately reduced ARSA activity had the ARSA-PD allele, which only lowers the enzyme activity but does not induce any phenotypic symptoms, even in patients with homozygous alleles [5, 6]. Two cases had the pathogenic allele c.302G>A, which is the most common mutation in the Japanese MLD patients [8].
Case No. 1 was homozygous for the c.1107+1delG allele. Therefore, we concluded that this was a novel pathogenic variant. This was verified by performing targeted mutational analysis of the patient's parents and subsequent cDNA analyses. Results of the genetic analysis were consistent with clinical findings such as spasticity; motor function abnormalities, including gait disturbance; and white matter disease confirmed by brain imaging. Kang et al. [9] reported a splicing mutation at the same intronic site. However, this mutation was a substitution change (c.1107+1G>T) and was different from the mutation identified in the present study.
Case No. 2 and No. 3 had two compound heterozygous ARSA-MLD alleles, which confirmed the diagnosis of MLD. The mutation c.292delinsCC in case No. 3 who was confirmed as having MLD was not present in the HGMD database and hence was termed as a novel pathogenic mutation. Moreover, these two cases also had ARSA-PD allele (c.1178C>G) which contributed to a reduction of ARSA activity [15].
Case No. 4 had one heterozygous ARSA-MLD allele (c.465+ 1G>A) and two heterozygous ARSA-PD alleles (c.1055A>G and c.1178C>G) [16]. In addition, this case had the c.585G>T allele, which reduced ARSA activity similar to the ARSA-PD allele [17]. The presence of these alleles in case No. 4 may explain the observed reduction in ARSA activity.
Case No. 5 was not diagnosed as having MLD because of the presence of only one heterozygous ARSA-PD (c.1055A>G) allele. This might explain the slightly lower ARSA activity in this individual.
Case No. 6 and No. 7 were the parents of case No. 1, and each had a heterozygous c.1107+1delG mutation, as confirmed by using targeted mutational analysis, indicating that they were obligate heterozygotes. cDNA analysis identified an altered splicing region in exon 6 of the variant allele that was 5 bp ahead of its position in the normal allele. ARSA activity in each parent was lower than the normal range, which was consistent with the results of genetic analysis.
In conclusion, of the seven subjects included in our study, three were confirmed as having MLD on the basis of genetic analysis. We also identified a novel pathogenic variant, c.1107+1delG, by performing familial and cDNA analyses. Total sulfatide concentrations were inversely correlated with ARSA activity. The results of this mutational and biochemical study on MLD will increase our understanding of the genetic characteristics of MLD in Koreans. In addition, these results indicate that gene mutational analysis can be used to confirm the diagnosis of MLD.
 XML Download
XML Download