

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Cancer-associated retinopathy (CAR) is a paraneoplastic neuronal degeneration in the retina of a cancer patient and is characterized by the rapid, progressive loss of vision [1]. The CAR has been described to occur with small-cell lung cancer, breast cancer, gynecological cancers, and others, in which the remote effects of cancers may stem from autoimmune responses of serum autoantibodies against both tumor and retinal proteins [2,3]. Pathologically, CAR is the degeneration of photoreceptors, with relative sparing of the inner retina, including ganglion cells, bipolar cells, amacrine cells, and horizontal cells, which are supplied by retinal vessels [1-3]. Although the precise mechanism has not been fully established, CAR may primarily result from autoimmune-mediated apoptosis, which is supported by the lack of inflammatory response to the retinal degeneration of CAR [4,5]. Among those autoantibodies found in sera of patients with CAR, those against the 23-kDa retinal protein recoverin appear to be the most common [1,6].
Recoverin is a cytoplasmic calcium-binding protein that is distributed in photoreceptor cells, bipolar cells, and in a small population of cells in the ganglion cell layer [7]. In CAR, it has been hypothesized that a high titer of autoantibodies are able to cross the blood-retinal barrier (BRB) and penetrate susceptible retinal cells, which is followed by cross-reactivity with particular retinal proteins [1,2]. Binding of the anti-recoverin antibody to recoverin enhances rhodopsin phosphorylation, continuously opens cGMP-gated channels, and increases the intracellular calcium level, which leads to activation of the caspase-dependent apoptotic pathway [5]. It has been already well-established that autoantibodies can enter and kill retinal cells [4,5], whereas it has never been established whether autoantibodies can cross the BRB [8,9]. In addition, although intravitreally injected autoantibodies successfully induced retinal degeneration and dysfunction similar to those of CAR [10,11], the intravitreal autoantibody injection is somehow different from the natural pathogenesis of CAR, in which it is hypothesized that circulating autoantibodies that cross the BRB enter retinal cells to induce cell death.
To maintain a stable environment in order to ensure the appropriate activities of retinal neurons, retinal vessels that supply the inner retina form the specialized structure of the BRB [11], which is regulated by modulating both angiogenesis and tight junction formation, as described in our previous findings [12,13]. The BRB is actually located in retinal endothelial cells, whose specific junction molecules are requisite for the maintenance of the barrier function [13,14]. In retinal endothelial cells, tight junction proteins such as zonula occludens (ZO)-1 and occludin are well-characterized, the expressions of which are inversely related to permeability in the BRB [13,15,16]. In a variety of retinal pathologies including uveoretinitis, diabetic retinopathy, or age-related macular degeneration, diverse mechanisms might affect the integrity of the BRB and lead to breakdown [13,14]. Although less is understood about increased vascular permeability and extravasation from the bloodstream in BRB breakdown, it has been proposed that either process may occur to allow for migration through intercellular endothelial tight junctions and for trans-cellular migration through endothelial cells [17,18].
In this study, we demonstrated that intravenously administrated anti-recoverin antibodies could not pass through the BRB and, therefore, do not cause retinal cell death. Moreover, even under the condition of BRB breakdown, anti-recoverin antibodies could not cross the BRB and remained on retinal vessels without retinal cytotoxicity.
Materials and Methods
Animals
Female C57BL/6 mice (eight- to ten-weeks-old, body weight 22 ± 1.6 g) were purchased from Samtako (Seoul, Korea). Care, use, and treatment of all animals in this study were in strict compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The mice were housed under standard 12-hour dark-light cycles at approximately 23℃ room temperature. After intravenous administration of anti-recoverin antibodies, the mice were sacrificed and enucleated at one and seven days. Each group contained ten animals. The enucleated eyes were immersion-fixed in 4% paraformaldehyde and subsequently embedded in paraffin.
Antibody
Anti-recoverin polyclonal antibodies (Chemicon International Inc., Temecula, CA, USA) were used in this study and were formulated in rabbit serum. According to the manufacturer's specifications, the antibody has specificity for recoverin with immunogenicity for recombinant human recoverin and reacts with mouse recoverin, as well as those of primates, including humans. Anti-recoverin antibodies (40 µL, 1 mg/mL) were intravenously administrated after dialysis to achieve systemic applications.
Cell culture
Human retinal microvascular endothelial cells (HRMECs) were purchased from the Applied Cell Biology Research Institute (Kirkland, WA, USA) and grown on attachment factor-coated plates in complete medium (Cell Systems, Kirkland, WA, USA). The HRMECs used in this study were taken from passages four to six.
Immunohistochemistry
Four-micron-thick serial sections were prepared from paraffin blocks. Sections were deparaffinized and hydrated via sequential immersion in xylene and graded alcohol solutions, treated with proteinase K for 5 minutes at 37℃, and then treated with normal serum obtained from the same species in which the secondary antibody was developed for 10 minutes to block nonspecific staining. To stain intravenously administrated anti-recoverin antibodies, slides were incubated with FITC-conjugated IgG (1:400; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and TRITC-conjugated IgG (1:400; Jackson ImmunoResearch, Philadelphia, PA, USA). To stain endothelial cells or astrocytes, slides were incubated overnight at 4℃ with rabbit polyclonal antibody against CD31 for endothelial cells (1:100, Santa-Cruz Biotechnology) or glial fibrillary acidic protein (GFAP) for astrocytes (1:100; Dako, San Francisco, CA, USA). FITC-conjugated IgG (1:400, Santa Cruz Biotechnology) and TRITC-conjugated IgG (1:400, Jackson ImmunoResearch) were used as secondary antibodies. For staining of nuclei, slides were stained with 10 µg/mL 4', 6-diamidino-2-phenolindole (Sigma-Aldrich Co., St. Louis, MO, USA). The slides were observed under a fluorescence microscope (BX50; Olympus, Tokyo, Japan).
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay
Four-micron-thick serial sections were prepared from paraffin blocks. Transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed with a kit (ApopTag; Intergen, Purchase, NY, USA), according to the manufacturer's instructions. TUNEL-positive cells were evaluated by two masked and independent observers (KJH and KJH) who viewed ten randomly-selected fields in each slide at a 400× magnification using a fluorescence microscope (BX50, Olympus).
Leakage assessment through the perfusion of retinal vessels with fluorescein-conjugated dextran
According to our recent study [16], one day following intravitreal injection of 20 ng/ml vascular endothelial growth factor (VEGF, Sigma-Aldrich Co.), deeply anesthetized mice were perfused through the tail vein with high molecular weight (MW= 500,000) fluorescein-conjugated dextran (Sigma-Aldrich Co.). At 1 hour post-perfuson, the eyes were enucleated and fixed in 4% paraformaldehyde for 2 hours. The retinas were dissected, flat-mounted in Dako mounting medium (DakoCytomation, Glostrup, Denmark), and viewed under fluorescence microscopy (BX50, Olympus) at 400×. These experiments were repeated at least three times.
Western blotting analysis
Western blotting was performed using standard Western blotting methods. The protein concentration was measured using a BCA protein assay kit (Pierce, Rockford, IL, USA). Equal amounts of protein were separated via electrophoresis on a 5% to 10% SDS-PAGE gel and transferred electrophoretically onto a nitrocellulose membrane (Amersham, Little Chalfont, UK). The membranes were blocked for 30 minutes in 5% non-fat milk. Next, the membranes were incubated overnight with anti-CD31 (Santa Cruz Biotechnology), anti-ZO-1 (1:1000; Zymed, San Francisco, CA, USA), anti-ZO-2 (1:2000, Zymed), and anti-occludin (1:1000, Zymed) at 4℃. After they were washed with phosphate balanced solution (PBS-T), the membrane was incubated for 1 hour at room temperature with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG (1:10,000; Pierce) in PBS-T and 1% non-fat milk. To ensure the equal loading of protein in each lane, the blots were stripped and reprobed with an antibody against β-actin. Intensity values were normalized relative to control values. The blots were scanned using a flatbed scanner, and the band intensity was analyzed using the TINA software program (Raytest, Straubenhardt, Germany).
Results
Intravenously administrated anti-recoverin antibodies could not pass through the BRB
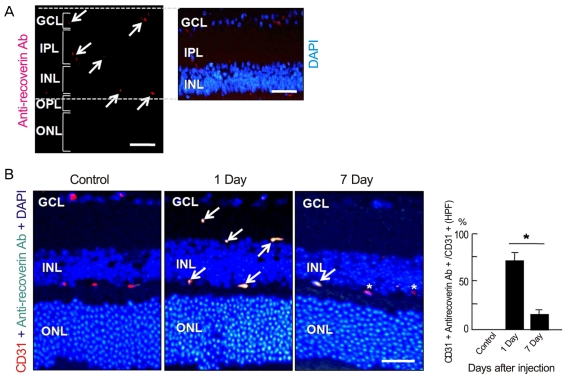
To investigate whether intravenously administrated anti-recoverin antibodies could cross the BRB, we injected 40 µg of anti-recoverin antibodies into the tail veins of C57BL/6 mice. As shown in Fig. 1A, anti-recoverin antibodies were exclusively detected in the inner retina, including the ganglion cell layer, inner plexiform layer, inner nuclear layer, and outer plexiform layer, whose distribution interestingly seemed to be confined to the retinal vessels.
To address the distribution of anti-recoverin antibodies on retinal vessels, we compared the distribution of anti-recoverin antibodies with the expression of CD31, an endothelial cell marker. Surprisingly, anti-recoverin antibodies were nearly co-localized with CD31 expression one day following injection, and the distribution pattern remained for seven days after injection, although much of the anti-recoverin antibodies disappeared from the retina (Fig. 1B).
Intravenously administrated anti-recoverin antibodies never induced cytotoxicity in the retina
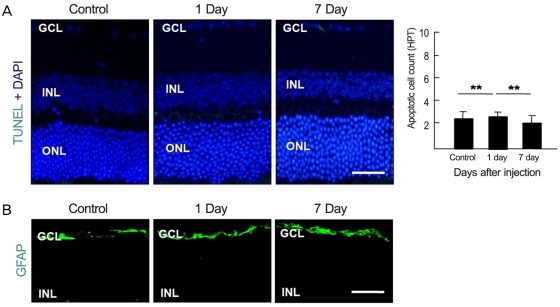
To examine whether intravenously administrated anti-recoverin antibodies could induce retinal cell death, based on the previous reports of apoptotic cell death induced by autoantibodies, we evaluated apoptotic cell death in the retina using a TUNEL assay. As demonstrated in Fig. 2A, intravenously administrated anti-recoverin antibodies never induced retinal apoptotic cell death, as would be expected if they could not cross the BRB. In addition, we investigated whether intravenously administrated anti-recoverin antibodies could up-regulate GFAP expression in the retina as an indicator of retinal stress [19]. The injection of anti-recoverin antibodies never induced a progressive increase in GFAP in Mueller cells (Fig. 2B).
Intravascular anti-recoverin antibodies could not pass through the BRB under breakdown conditions
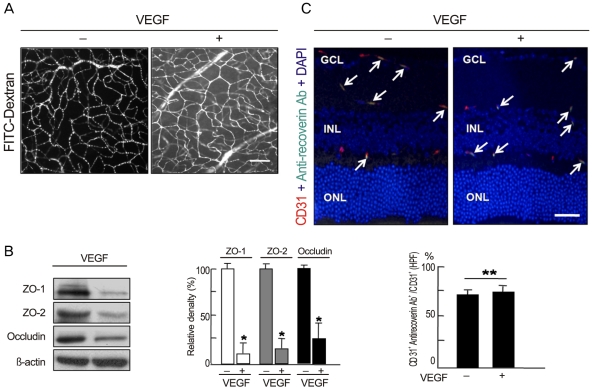
To evaluate whether intravenously administrated anti-recoverin antibodies could pass through the BRB under the conditions of BRB breakdown, we induced VEGF-mediated BRB breakdown via intravitreal injection of 20 ng/mL VEGF and analyzed the passage of autoantibodies across the BRB. Intravitreal injection of VEGF disrupts the BRB and induces profuse leakage from retinal vessels (Fig. 3A), which is closely linked to a decrease in tight junction proteins in retinal endothelial cells (Fig. 3B) [14,18]. Even during BRB breakdown, intravenously administrated anti-recoverin antibodies were confined to retinal vessels without passage across the BRB, were co-localized with CD31, and were detected on retinal vessels with a similar frequency to that of the control (Fig. 3C).
Intravenously administrated anti-recoverin antibodies under BRB breakdown conditions never increased retinal cell death
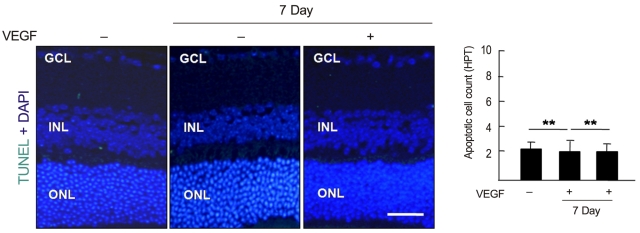
To examine whether intravenously administrated anti-recoverin antibodies under BRB breakdown conditions could induce retinal apoptotic cell death, a TUNEL assay was performed. As demonstrated in Fig. 4, there was no significant change in apoptotic cell count with or without BRB breakdown.
Discussion
CAR has been thought to be mediated by the autoimmune reactions triggered by antigenic mimicry, where anti-recoverin antibody plays a crucial role in the pathogenesis of CAR, resulting in retinal degeneration [2,4-6]. It is hypothesized that autoantibodies against recoverin develop from a tumor antigen resembling one shared by recoverin, and a high titer of circulating antibodies may increase the possibility of passage through the BRB. Although retinal degeneration in CAR may be mediated by both humoral and cellular immune mechanisms [20], it is not clear whether either plays a major role. As proposed in other autoimmune diseases, anti-recoverin antibodies are able to enter into retinal cells and induce apoptotic cell death, which has been demonstrated in retinal tissue in vivo as well as in an in vitro retinal cell culture [4,5,9,10]. However, we still do not know how anti-recoverin antibodies pass through the BRB because anti-recoverin antibodies were intravitreally injected in the in vivo experiments.
In this study, we demonstrated that intravenously administrated anti-recoverin antibodies could not pass through the BRB. Based on the high titer of autoantibodies in CAR patient serum samples [1,2], it has been hypothesized that a high titer of anti-recoverin antibodies is consequently related to penetration through the BRB. In addition, cancer patients with a low titer of anti-recoverin antibodies were reported to demonstrate no visual dysfunction [8]. Given that the blood volume in mice is about 85 to 95 mL/kg [21], and the body weight of the mice used in our experiment was 22 ± 1.6 g, the amount of intravenous injection, 40 µg, is a relatively high titer compared to the serum concentration of CAR patients [1,2]. However, anti-recoverin antibodies were exclusively observed in the retinal vessels. Interestingly, anti-recoverin antibodies still remained on retinal vessels for seven days after intravenous administration, although the frequency decreased. Furthermore, intravenous administration of anti-recoverin antibodies never induced any retinal stress, confirmed by a lack of change in GFAP expression, as well as no retinal cell death. It is reasonable to speculate that these results occurred as a result of no anti-recoverin antibodies crossing the BRB.
Based on retinal vascular changes indicating BRB breakdown in CAR [22], the pathogenesis of CAR might be linked to BRB breakdown. The BRB is a selective barrier that represents the boundary between the systemic circulation and the retinal neuron. Vascular permeability in the retina is therefore actively regulated by the BRB, which is established with a tight junction restricting the paracellular flux of molecules [12,13,18]. VEGF is causally linked to BRB breakdown through the dysregulation of the tight junction, which leads to increased retinal vascular permeability [13,18]. In addition, VEGF upregulates intracellular adhesion molecule (ICAM)-1 in retinal endothelial cells, which increases leukocyte adhesion [23].
The ICAM-1 in endothelial cells activates a Rho A-mediated signal to allow leukocytes to paracellularly migrate across the barrier through the interendothelial cell junction or transcellularly through the endothelial cell itself [17]. In this study, we induced BRB breakdown via intravitreal injection of VEGF, which might enhance penetration of intravascular anti-recoverin antibodies into the retina. Surprisingly, there was no passage of anti-recoverin antibody through the BRB in the case of VEGF-induced BRB breakdown. In addition, anti-recoverin antibodies were observed on retinal vessels with a similar frequency to that of the control, which in part supports the idea that penetration of anti-recoverin antibodies through the BRB might not be mediated by regulation of the tight junction in restriction of paracellular flux, although this is not certain for the transcellular route. The interpretation of this result is only speculative and requires further investigation.
In conclusion, intravenously administrated anti-recoverin antibodies could not pass through the BRB, which therefore does not induce either retinal stress or retinal cell death. Interestingly, intravascular anti-recoverin antibodies could not migrate into the retina even in VEGF-induced BRB breakdown, which also was not accompanied by an increase in retinal cell death. On the basis of the available evidence, we suggest that intravascular anti-recoverin antibodies could not naturally penetrate into the retina, and BRB breakdown mediated by dysregulation of the tight junction might not be sufficient to allow anti-recoverin antibodies to pass through the BRB. Further investigations are required to elucidate other transporting routes of intravascular anti-recoverin antibodies to the retina and for the sustained existence of intravenous administrated anti-recoverin antibodies on retinal vessels.
 XML Download
XML Download