

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Over 150 cases of Hallermann-Streiff syndrome (HSS, oculo-mandibulo-dyscephaly with hypotrichosis) have been reported worldwide since it was first described as HSS by Francois in 1958 [1,2]. This entity has a typical physical appearance: bird-like facies, abnormal dentition, hypotrichosis, skin atrophy, proportionate dwarfism, and ophthalmic features such as congenital cataracts and microphthalmia [3-15].
To our knowledge, few reports have addressed surgical correction of ptosis with strabismus in HSS. We report a patient diagnosed with HSS who had several uncommon ocular manifestations [4,5]-esotropia, entropion, and blepharoptosis, and we describe successful surgical correction of this condition.
Case Report
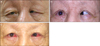
A 54-year-old man visited Yeouido St. Mary's Hospital complaining of ocular discomfort due to cilia that had been touching the corneas of both eyes for several years. The patient had been treated for intraocular hypertension with anti-glaucoma preparations. There was no other notable family history. He had bird-like facies, a pinched nose, hypotrichosis of the scalp, mandibular hypoplasia with forward displacement of the temporomandibular joints, a small mouth with abnormal dentition, and proportional short stature (about 150 cm in height). His general appearance led us to diagnose him with Hallermann-Streiff syndrome (Fig. 1). His right visual acuity was 0.04 (0.1 × -2.75 Ds = -3.0 Dc A × 90°) by Snellen chart, and his left visual acuity allowed him to count fingers at 20 cm (uncorrected × -2.5 Ds = -1.25 Dc A × 90°). Keratometry measurements were K1 = 57.00 / K2 = 58.75 in the right eye and K1 = 54.50 / K2 = 55.75 in the left eye. His axial length was 23.77 mm in the right eye and 23.69 in the left eye. Ophthalmic features included sparse eyelashes and eyebrows, microcornea (cornea diameter of right eye vertical/horizontal (V/H) = 9.25/8.75 mm, left eye (V/H) = 9.0/8.75 mm), nystagmus, right lower eyelid entropion, and bilateral upper eyelid entropion with aponeurotic blepharoptosis. Right marginal reflex distance 1 (MRD1, distance from upper eyelid margin - corneal reflex) was 1 mm, left MRD1 was 0 mm, and levator palpebrae function was 10 mm in both eyes. We noted lid drooping at his inferior gaze and an overactive frontalis muscle compensating for weak levator function, especially on the right side. There was esodeviation of the eyeball of more than 100 prism diopters at near and distance. Limited ocular movement was present on lateral gaze (Fig. 2). Both corneas were clear but showed some superficial erosions caused by cilia irritation. The anterior chambers were deep. The patient had never undergone cataract surgery, but he was aphakic in both eyes. Fundus examination showed pale optic discs and severe chorioretinal atrophy in both eyes. The patient refused to undergo DNA analysis or imaging evaluation for other associated systemic findings, such as spina bifida, scoliosis, lordosis, respiratory disorders, and dental abnormalities.
The capsulopalpebral fascia was repaired to treat right lower eyelid entropion, but an additional Quickert suture was needed to prevent recurrence. Upper eyelid blepharoplasty and levator palpebrae repair were performed for correction of aponeurotic blepharoptosis and dermatochalasis. We noted very thin eyelid skin and tarsal plates, fatty infiltration in front of the tarsal plates, and severe fat tissue replacement of the levator palpebrae. Frontalis muscle overactivity was relieved after the correction of eyelid problems (Fig. 3B). Three months after the eyelid surgeries, the right medial rectus was recessed 7.5 mm, the left medial rectus was recessed 7.25 mm, and the left lateral rectus muscle was resected 8.0 mm under general anesthesia with endotracheal intubation (Fig. 3C). Biopsy of the resected muscles revealed atrophy and chronic perivascular infiltration of inflammatory cells. The patient had persistent esodeviation of about 20 prism diopters but had no other lid complications at his last visit.
Discussion
The etiology and pathogenesis of HSS remain unknown. Most cases seem to be sporadic, but some genetic abnormalities and family histories have been reported [1,9]. HSS patients exhibit many abnormalities involving various systems: craniofacial, mandibular, and dental anomalies with or without musculoskeletal, cardiac, or respiratory problems and mental retardation [1-3,7-10]. Diagnosis is based on clinical findings like dyscephaly with bird-like facies, abnormal dentition, hypotrichosis, atrophy of the skin (especially on the nose), congenital cataracts, bilateral microphthalmia, and proportionate dwarfism [3]. Ocular abnormalities are a major problem, with the most common ocular features being microphthalmia and cataracts, which are present in 90% of HSS patients [1,2,6-8].
Seven cases of HSS have been reported in Korea: 2 by pediatricians and 5 by ophthalmologists [9,12-17]. Although congenital cataracts are one of the most common characteristics of HSS, two previously reported patients had spontaneous aphakia [9,15]. The patient in this report had no notable family history, cardiac or respiratory disorders, or mental retardation.
Table 1 shows the ophthalmic manifestations that have been reported in patients with HSS, as well as the positive findings in this case [3-11]. The patient had normal range of axial length, but did have microcornea, which is uncommon in HSS. He was also aphakic, despite the fact that he had never undergone cataract surgery. The lenses may have been absorbed spontaneously after birth as he advanced in age, which sometimes occurs in the setting of HSS [1,9,15]. Other ophthalmic findings included nystagmus, intraocular hypertension, sparse eyelashes and eyebrows, esotropia, and eyelid problems: blepharoptosis and entropion. To our knowledge, entropion is especially uncommon in HSS, and few cases have been reported. We performed entropion and blepharoptosis repair after administering a local anesthetic injection. During eyelid surgery, we noted very thin skin. Hence, any manipulation should be done carefully. The patient's precise esodeviation could not be determined because of his nystagmus, but it was certainly over 100 prism diopters. Considering the preoperative extraocular muscle limitations, we chose 3 muscles to reposition no more than 8 mm. Because of poor visual acuity, the patient had no diplopia before or after surgery.
One of the most severe complications in HSS is respiratory embarrassment. David et al. [1] reported that general anesthesia with endotracheal intubation was difficult and risky in HSS, even in its relatively mild form. Many cases of respiratory distress have been reported, especially in the postoperative period. In the current case, we prepared by consulting the anesthesia team before strabismus surgery. Fortunately, the patient recovered from anesthesia without any respiratory complications.
In conclusion, we present a case of successful surgical repair for HSS in a patient who had no complications but who had uncommon ophthalmic features of strabismus, entropion, and blepharoptosis. Careful anesthetic and postoperative management are needed in the setting of HSS.



 XML Download
XML Download