

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Activated protein C is an anticoagulant enzyme that inactivates the plasma factors Va and VIIIa by limited proteolysis, thereby inhibiting the conversion of factor X to factor Xa and of prothrombin to thrombin. It also has been reported to induce a stimulatory effect on fibrinolytic activity.1
Protein C deficiency is an autosomal recessive disorder. Heterozygous individuals may occasionally have an increased disposition to recurrent venous thrombosis or more usually may remain asymptomatic and have protein C levels of approximately 50%. Homozygous individuals may develop widespread life threatening thromboses involving the central nervous system, eyes, kidneys, and skin in the neonatal period, usually after an uncomplicated, full-term pregnancy and delivery.2 Protein C activity is usually less than 1% (normal 70-140%).3 Homozygous protein C deficiency is very rare with an estimated incidence of one in 500,000 to one in 750,0004 and ophthalmic findings such as vitreous hemorrhage, retrolental membrane,5 a dense, funnel shaped mass suggesting persistent hyperplastic primary vitreous (PHPV),5-7 retinal arterial or venous occlusion,8 and subretinal hemorrhage2 have been reported. We present a rare case of a neonate with homozygous protein C deficiency who had bilateral retinal dysplasia and secondary glaucoma, and who underwent bilateral lensectomy to save the eyeballs.
Case Report
A female neonate was born weighing 3.4kg after an uneventful pregnancy at 42+1 weeks of gestation. Family history was unremarkable except the mother had previously had a stillborn daughter. There was no known consanguinity between the parents. On day 2, areas of purpura developed on her right thigh, ankle, and buttocks. The purpura grew larger, skin necrosis progressed and she was referred to the neonatal intensive care unit. Prothrombin time was 14.3 seconds, partial thromboplastin time was 36.2 seconds, fibrinogen level was 95.3 mg/dl (decreased), platelet count was 108,000/mm3, fibrin-gradation products were >160 µg/ml (normal, <10 µg/ml) and screening for TORCH was negative. On the presumed diagnosis of disseminated intravascular coagulation, she was started on replacement therapy with fresh frozen plasma (FFP). Skin biopsy result was consistent with purpura fulminans. Further evaluation disclosed low levels of protein C activity (10%) and protein C antigen (<12%). Both parents and a 4-year-old elder sister showed reduced levels of protein C activity (71%, 73% and 63%, respectively). On the basis of these results, a diagnosis of homozygous protein C deficiency was made. Both parents and the sibling were presumed to be heterozygous for the deficiency.
On day 2, bilateral leukocoria was noted by the parents and ophthalmic examination on day 5 in another hospital showed diffuse corneal opacity and shallow anterior chamber in both eyes. Both eyes were microphthalmic and posterior synechiae with pupillary membrane were present. On day 7, B-scan ultrasonography revealed decreased anterior chamber depth, microphthalmos and vitreoretinopathy suggesting PHPV in both eyes. The patient remained stable under intravenous FFP therapy and additional oral administration of warfarin.
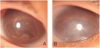
The patient was firstly referred to our institute on day 41 with the referral diagnosis of PHPV. Ophthalmic findings were similar to those at day 5 except for the absence of microphthalmos. In both eyes, intraocular pressures (IOP) by pneumatic tonometer was 14mmHg, anterior chamber depth was extremely shallow and corneal diameter was 10mm. The axial length of the right and left eyeballs was 17.10 mm and 16.75 mm, respectively. Computed tomographic scan showed a high attenuated lesion posterior to the vitreous body. B-scan ultrasonography, which revealed decreased anterior chamber depth, intravitreal mass lesions without microphthalmos and a funnel-shaped retinal detachment, suggested bilateral retinal dysplasia (Fig. 1).
To treat the progression of corneal opacity due to shallow anterior chamber, bilateral lensectomies through pars plicata entry were performed simultaneously on both eyes on day 69 under continuous replacement therapy with FFP. During the operation, fibrovascular aggregations which could not be removed were observed in the vitreous cavity, but elongation of ciliary process was not observed. Lensectomy was done with the vitrectomy cutter, simultaneous with anterior chamber formation. The patient showed good postoperative hospital course and an absence of any early postoperative complication. At 6 months old, she displayed decreased corneal opacity and deep anterior chamber in both eyes. On her last follow-up at 14 months, she did not show any signs of light perception. Slit lamp examination showed clear cornea and deep anterior chamber in her right eye, but the left eye showed moderate corneal opacity with a band keratopathy. However, the patient did not show phthisis bulbi or retardation of bony orbital growth (Fig. 2).
Discussion
The definite diagnosis of homozygous protein C deficiency is difficult in the neonate because the concentrations of the vitamin K-dependent coagulation factors are reduced in comparison with adult levels.2 The protein C activity level of the present patient (10%) was not as low as that of other cases of reported homozygous protein C deficiency, which were usually less than 5%.2,5-7,9 Considering the moderately decreased levels of protein C activity of both parents and the sibling, our patient was diagnosed as homozygous type. The mother had earlier had a stillbirth daughter who had hydrocephalus and oligohydroamnios. Considering that some cases of homozygous protein C deficiency had hydrocephalus,5,6 the child was presumed to be homozygous type, too.
The first signs of protein C deficiency have been detected from shortly after birth to 2 months after birth.2,5 Ophthalmic manifestations include leukocoria, nonreactive pupil,2 periorbital edema, shallow anterior chamber,2,5,6 and microphthalmos. Rarely, leukocoria by itself can be the first manifestation of homozygous protein C deficiency, even before any other systemic manifestations like purpura fulminans.9 Ophthalmic manifestations of homozygous protein C deficiency usually result from abnormalities of the posterior segment,2,5-9 and in the further course, retinal detachment develops as a result of these conditions in many cases.2,5,8 One or both eyes may be affected.
Although the patient was diagnosed as PHPV in the previous hospital, vitreoretinopathy in both eyes suggested retinal dysplasia rather than PHPV. The patient's status could not be described as microphthalmos because the corneal diameter and axial length of the eyeballs at 41 days old were within the normal range of development, and bilateral aggregation of fibrovascular material regarded as retina tissue further suggested the diagnosis of retinal dysplasia. Above all, we could not observe any evidence of ciliary process elongation, which is typical of PHPV, during surgery. Norrie disease was suspected as another possibility, and we recommended DNA analysis for Norrie disease gene or chromosomal study to find other systemic abnormalities like trisomy 13. The parents, however, refused.
A few cases of homozygous protein C deficiency showing a shallow anterior chamber have been reported.2,5,6 In addition to that, our patient showed progressing bilateral corneal opacity and was, despite IOP measured by pneumatic tonometer being 14 mmHg in both eyes, thought to be under the progression of bilateral secondary pupillary block angle closure glaucoma developed due to progressing vitreoretinopathy. In glaucoma combined with retinal dysplasia, the rise in IOP is often transient and not marked.10 On this occasion, the clues for diagnosis should be corneal opacity and shallow anterior chamber, rather than increased IOP. Shortly after bilateral lensectomy, corneal opacity was regressed to some degree and the eyeballs were saved until 14 months old, 1 year after the surgery. Nevertheless, at that time, newly developed band keratopathy with remaining moderate corneal opacity in the left cornea was observed. Shallow anterior chamber suggested corneal decompensation due to progressing vitreoretinopathy, despite the lensectomy.
To the best of our extent of knowledge, this is the first Korean report of ophthalmic involvement and its surgical treatment in homozygous protein C deficiency. Up to now, ophthalmic surgery in homozygous protein C deficiency has been reported only in one case worldwide. Hermsen et al6 described a girl who underwent pars plana vitrectomy and lensectomy in an eye with a dense, funnel shaped retrolental mass that was suggestive of PHPV to open the optic axis when she was 17 days old. At 3 years after surgery, ophthalmic examination showed light perception in the eye. Dreyfus et al11 reported unilateral or bilateral blindness in six of nine children with severe congenital protein C deficiency. Similarly, our patient did not show any light perception at 14 months old. Although visual outcome was very poor after surgery, we could impede or slow down the progression of secondary glaucoma and save the eyeballs in the infant with homozygous protein C deficiency. Any progressing vitreoretinopathy can result in secondary angle closure glaucoma. Detailed ophthalmic examination is important for the early detection of signs of secondary glaucoma and the consequent chance of saving the eyeballs in a neonate with homozygous protein C deficiency.

 XML Download
XML Download